The Promise of NK Cell Therapeutics
Introduction
![]() NK cells are the first line of defense against cancer and have been in the spotlight as a cancer therapy in the last decade. Their advantages versus T cells, such as their safety profile and not requiring MHC (Major Histocompatibility Complex)-antigen stimulation to become cytotoxic, have made NK cells attractive. However, as with T cells, the TME (tumor microenvironment) can suppress their activity, and infiltration into solid tumors is still a challenge. In recent years, research has focused on the development of strategies that allow NK cells to sustain cytotoxic function, increase residence time in vivo and methods to process and efficiently expand them ex vivo. We will introduce NK cell biology, discuss challenges and progresses made in the NK immunotherapy field and introduce innovative products by BPS Bioscience that support the field to move forward.
NK cells are the first line of defense against cancer and have been in the spotlight as a cancer therapy in the last decade. Their advantages versus T cells, such as their safety profile and not requiring MHC (Major Histocompatibility Complex)-antigen stimulation to become cytotoxic, have made NK cells attractive. However, as with T cells, the TME (tumor microenvironment) can suppress their activity, and infiltration into solid tumors is still a challenge. In recent years, research has focused on the development of strategies that allow NK cells to sustain cytotoxic function, increase residence time in vivo and methods to process and efficiently expand them ex vivo. We will introduce NK cell biology, discuss challenges and progresses made in the NK immunotherapy field and introduce innovative products by BPS Bioscience that support the field to move forward.
NK Cells
NK cells derive from CD34+ hematopoietic stem cells and can be found in the blood, where they account for 5-15% of the lymphocyte population, liver, uterus and spleen. NK cells are a heterogeneous population of cells at different stages of maturation, a process regulated by multiple factors, such as Eomes (T-box brain protein 2) and BLIMP-1 (B lymphocyte-induced maturation protein 1). In humans they can be identified by the absence of CD3 and presence of CD56, with CD56 and CD16 expression levels correlating with their functional properties. The CD56brightCD16low population accounts for less than 15% of the total and represents a more immature stage, being present mostly in tissues like tonsils and lymph nodes. A highly cytotoxic subset of NK cells is characterized by CD56dimCD16high (1, 2). CD56dim cells contribute to fight cancer by being highly cytotoxic cells, while CD56bright (also known as NKreg) contribute by having the capability to release cytokines and chemokines when stimulated by IL-12, IL-15 and IL-18, and promote maturation and activation of dendritic cells, macrophages and T cells. Of note, there is a subset of CD56+ cells that also has CD3 expression, the NKT (Natural Killer T) cells, which should be considered when relying only on CD56 as selection marker and can have an impact on clinical outcomes (3).
NK cells belong to the innate immune system, meaning that they do not require a memory related trigger to kill abnormal cells. Their function is tightly regulated by the balance of activating and inhibitory receptors belonging to a number of receptor families, including the natural cytotoxic receptor family (NCR), natural killer group 2 calcium dependent lectin family (NKG2), nectin-binding adhesion molecules and killer cell immunoglobulin like receptor (KIR) family (Table 1).
Table 1: NK activating and inhibitor receptors and respective ligands (1, 2, 4).
| NK Activating Receptor | Ligands |
|---|---|
| NKG2D | -MICA/B (MHC class I chain related protein A/B)-ULBP1-6 (UL16-binding protein) |
| DNAM1 (DNAX Accessory Molecule -1, or CD226) | -CD112 (nectin-2)-CD155 (PVR or poliovirus receptor) |
| CD16 (or FCγRIII) | -IgG Fc region |
| NKp46 (or NCR1, natural cytotoxicity receptor 1) | -Complement factor P -Vaccinia virus HA (hemagglutinin) -HS GAGs (heparan sulfate glycosaminoglycans) -Vimentin -Valreticulin on ER-stressed cells |
| NKp44 (or NCR2) | -HS GAGs -Influenza HA -PDGF-DD (platelet-derived growth factor DD) -Nidogen-1 -PCNA (proliferating cell nuclear antigen -Envelope proteins from West Nile and Dengue viruses |
| NKP30 (or NCR3) | -HS GAGs -Vaccinia HA -B7-H6 -Galectin-3 |
| NK Inhibitory Receptors | Ligands |
|---|---|
| CD94/NKG2A | -HLA-E |
| KIR2DL1 (killer cell immunoglobulin-like receptor 2DL1) | -HLA-C group 2 |
| KIR2DL2/3 | -HLA-C group 1 |
| KIR3DL1 | -HLA-Bw4 |
| KIR3DL2 | -HLA-A3, A11 |
| LIR-1 (leukocyte Ig-like receptor 1) | -HLA-G |
| TIGIT (T-cell immunoreceptor with IgG and ITIM domains) | -CD155 |
| LAG3 (lymphocyte-activation gene 3) | -MHC class II |
| PD-1 (programmed cell death protein 1) | -PD-L1 (programmed cell death ligand 1) |
Normal cells express MHC class I molecules on their surface allowing “licensing” to occur, where naïve NK cells are educated to recognize self and remain quiescent by the binding of HLA to KIRs. When HLA expression becomes deficient a “missing self” response occurs and the inhibitory brake is removed. NK cells use several mechanisms to destroy cancer cells, ranging from the initial direct release of lytic enzymes to a later death ligand-linked apoptosis, and indirect action via stimulation of other immune cells (Figure 1).
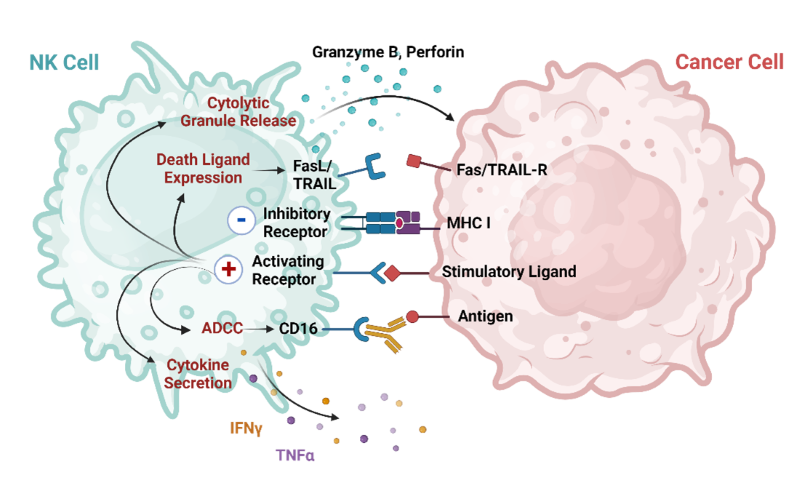
Figure 1: NK cell mechanisms of cancer cell destruction. Adapted from Du, N. et.al. (1).
In cancer cells MHC class I molecules are downregulated in order to avoid CD8+ T cell attack and end up releasing the contact with inhibitory receptors on NK cells. This triggers cytotoxicity responses via the direct release of perforin and granzyme. Typically, cells express low levels of KARs (killer activating receptors), however the genome instability of cancer cells can lead to overexpression of NKG2D and DNAM-1 ligands. KARs can trigger the production and release of “death ligands” such as TNFα (tumor necrosis factor alpha), FasL (Fas ligand) and TRAIL (TNF-related apoptosis-inducing ligand), triggering apoptosis and cell death. They also participate in immune regulation via the secretion of cytokines and chemokines involved in adaptive immunity activation, such as IFNγ (interferon gamma), IL-10, CCL3-5 (chemokine ligand 3-5) and lymphotactin. Another mechanism that NK cells use is through CD16, which triggers ADCC (antibody-dependent cellular cytotoxicity) when crosslinked with the Fc region of an antibody present in the cancer cell surface (1, 2, 3).
NK cell action is quick, with the production of cytokines occurring within minutes of stimulation, in contrast with the more delayed response of T cells, and highly active NK cells can restart the activation cycle and target 3 more cells, in a serial killing process (3). NK biology makes them efficient and perfectly adapted to be the natural killer patrol agents of the human body.
NK Cells and Cancer
NK cell functionality closely relates to cancer development, as demonstrated by individuals with a lower NK cytotoxicity profile being more susceptible to developing the disease. While NK cells have a range of tools available to eradicate cancer cells, these abnormal cells have also evolved mechanisms to resist attack.
Cancer cells can create an immunosuppressive TME towards NK cells and these suppressed NK cells are not only unable to fight cancer but become themselves suppressers of other anti-cancer responses. One strategy tumorigenic cells have developed involves MICA. MICA can be cleaved by ADAM (a disintegrin and metalloproteinases) proteins, which reduces the chance of cancer cells being recognized by NK cells but also allows the newly soluble MICA to block NKG2D from binding to MICA positive cells. Blocking ADAM proteins can be a potential strategic therapeutical option. Active TGFβ binds to NK cells and limits secretion of IFNγ, downregulates NKG2D expression, represses mTOR, inhibits glycolytic metabolism and suppresses proliferation and granzyme production. Cancer cells can also upregulate HLA molecules, FAS and PD-L1 and release PGE2, IDO, IL-6 and other NK cells inhibitors. In addition, cancer cells and immune cells compete for metabolites such as glucose and glutamine, creating an acidic and hypoxic TME, which decreases IFNγ secretion. The presence of immunosuppressive cells in the TME, such as Treg, macrophages, myeloid-derived suppressor cells and cancer associated fibroblasts serve to release immunosuppressive metabolites and to compete or use misleading decoys to inhibit interaction of NK cells with cancer cells. For instance, Treg cells can secrete vesicles containing IL-37, which works in multiple ways: it binds to IL-18Rβ, inhibiting the formation of IL-18 receptor complex and production of IFNγ, and binds to the negative checkpoint IL-1R8 (SIGRR or single immunoglobulin interleukin-1 related receptor). Overexpression of ligands of the KIRs TIGIT, CD96, LAG3 and PD-1 in cancer can also result in NK exhaustion (1, 3).
The understanding of the mechanisms involved in NK cell immunosuppression reveals the complexity of the interaction of the TME with immune cells but also opens opportunities to develop methods to counteract these mechanisms, some of which are currently being tested as cancer therapies.
NK and CAR-NK as Cancer Therapy Tools
NK cell-based therapy first showed its promise during hematopoietic stem cell transplants, where it was found that NK cells showed a graft versus leukemia effect. Several clinical trials using NK cells alone or in combination with other strategies are ongoing and showing promising results, with the current efforts in the field focusing on enhancing their efficacy.
The natural mechanisms of action of NK cells have been used to improve NK activity. The use of CD16 to trigger ADCC led to the development of antibodies able to recognize cancer antigens, resulting in higher NK cell tumor infiltration. Blocking NKG2A and LIR-1 increases cytotoxicity of NK cells and has shown results in the treatment of AML (acute myeloid leukemia) and ALL (acute lymphoblastic leukemia) (1). In addition to its natural mechanism, there are several ways that NK cells have been used or manipulated to treat cancer patients, as follows:
- Autologous NK Cell Infusion
In theory, the use of autologous NK cells presented itself as a feasible approach, as it bypassed the need for immunosuppression. However, by the time NK cells were to be collected the patients had undergone several treatments that impacted the expansion and cytotoxicity of the cells. Thus, other options required exploring.
- Non-Engineered Allogeneic NK Cell Infusion
One alternative to the use of autologous cell infusion is the use of allogeneic NK cells. Adoptive cell transfer seems to require that a lymphopenic environment is created so donor cells can survive and proliferate, but the results in hematological disorders are encouraging.
NK cells can be sourced from peripheral blood (PB), umbilical cord blood (UCB), or derived from NK immortalized cells line or iPSC (induced pluripotent stem cells). Each cell source has its benefits and downsides (Table 2). The ability to produce PB and UCB derived NK cells at clinical scale and how to retain their viability, expansion potential and cytotoxicity are challenges currently being addressed. The derivation of NK from iPSC is technically complex but holds the promise to be a real “off the shelf” out-patient therapy.
Table 2: NK cell sources and their main benefits and issues.
| NK Cell Source | Main benefits and issues |
|---|---|
| Peripheral Blood | -Easy to collect.-Heterogeneous population. -Low % of NK cells present and can result in one single dose. -Hard to genetically modify. |
| Umbilical Cord Blood | -Heterogeneous population.-Progenitor cells present at higher %. -% of overall NK cells is higher than in PB. -Hard to genetically modify. |
| NK92 | -Unlimited expansion. -High cytotoxicity. -Easier to manipulate. -Require irradiation prior to infusion. -Safety concerns. -Proprietary to ImmunityBio Inc. |
| iPSC | -Unlimited source of cells. -Requires differentiation into CD34+ progenitor cells, and then along the hematopoietic lineage, a process that is lengthy and technical challenging. -Easy to genetically modify. -Results in a homogeneous population. |
- Engineered Allogeneic NK Cell Infusion
While holding great promise, recent years have shown that CAR (chimeric antigen receptor)-T cell therapy has major drawbacks: it can induce cytokine release syndrome, GvHD (graft versus host disease) can occur, cells have minimal expansion and the length of time required to generate them is often incompatible with the patient lifespan and they seem to be inefficient in treating solid tumors. CAR-NK cells present themselves as a highly attractive option. NK cells produce less pro-inflammatory cytokines, allogeneic cells can be used without GvHD being a concern, and they can still target cancer cells even if the CAR target has been downregulated. Their life span is about 2 weeks, but persistence can be accomplished up to 12 months.
The CAR constructs used are similar to the ones used in CAR-T, with one single chain fragment (scFv) against the target of interest (CD19, HER2 and mesothelin are still the main targets) and a signaling domain composed of a fusion of CD3ζ with co-stimulatory molecules. While CD28-4-1BB can be used, the use of native signaling molecules such as NKG2D-2B4 can result in superior results (1, 2). Often a transgene encoding IL-15 is included. For example, the clinical trial involving NK cells expressing anti-CD19, IL-15 and an inducible caspase 9 CAR in patients with refractory CLL and non-Hodgin’s lymphoma is ongoing. 14 months post treatment 7 out of 11 patients had complete response, infused cells persisted up to 12 months post-infusion, cells seemed to home in the disease sites, and no cytokine storm syndrome or neurotoxic effects were seen (3, 5).
One interesting approach may be the use of both CAR-T and CAR-NK therapies at different time points during treatment. CAR NK cells would be infused first and would reduce immunosuppressive cells and secrete pro-inflammatory cytokines that could help improve the infiltration of CAR-T cells (2).
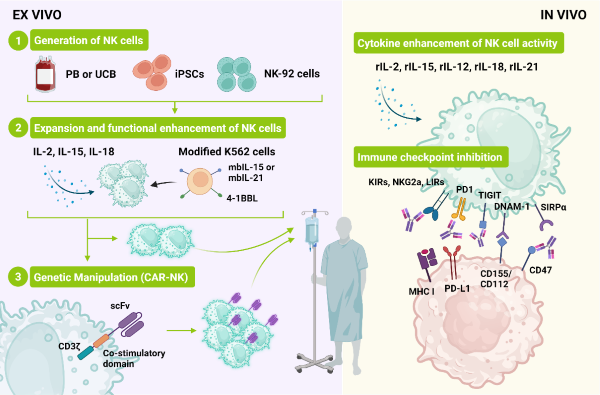
Figure 2: Sources of NK cells and methods to expand and increase their anti-cancer activity. NK cells can be derived from several sources and expanded in feeder-free (cytokines only) or feeder-containing conditions. CAR-NK cells can be generated and administered alone or in combination with priming (cytokine infusion) and inhibitors targeting NK immune-checkpoints. Adapted from Du, N. et.al. (1).
- Use of Killer Cell Engagers (BiKEs and TriKEs)
Bi and tri-specific killer engagers (BiKE and TriKEs) combine 2 or 3 single chain antibody variable fragments with different antigen specificities with the goal of bringing NK cells and cancer cells together, promoting the formation of immune synapses and ADCC. The targets most studied include CD19, CD20, CD33, HER2, EGFR and EpCAM. GTB-3550, a TriKE made of anti-CD16/IL-15/anti-CD33 proved the ability of this strategy to target CD33, maintain in vivo persistence, and it is now in clinical trials for hematological cancers (2).
- NK Cell Priming
The infusion of patients with cytokines, before or during NK cell infusion, such as IL-2, has been approved in the clinic, however safety concerns were raised. Efforts have been put into addressing side effects derived from those infusions and potentiating NK action.
IL-2 has affinity for IL-2Rα (CD25), which is present in Treg, and administration results in side effects due to activation of vascular endothelial cells. New molecules such as “super 2” with increased affinity for IL-2Rβ, and a fusion protein composed of IL-2 and OMCP (orthopoxvirus major histocompatibility complex class I like protein) have been created. IL-15, which is critical for maintaining NK cell proliferation, maturation and functionality, has been approved for use in refractory solid tumors, but likely because the patients have been heavily pretreated the results have not been as expected. Pre-activation with heterodimeric IL-15 (ALT-803), composed of IL-15 and IL-15Rα, was shown to increase cytotoxicity and ADCC in vitro and in a mouse model of lymphoma (2). N-803, an IL-15 superagonist made of mutant IL-15 (N72D) bound to IL-15Rα-IgG1-Fc, has increased affinity for IL-15Rβ resulting in higher biological activity and half-life (1).
Priming with a leukemia cell line CTV-1 is also being explored and a clinical trial using CTV-1 lysate primed NK cells is ongoing.
- Solid Tumor Targeting Strategies
Despite all these strategies targeting solid tumors is still a challenge, and bypassing the immunosuppressive TME, homing and NK cell infiltration are being intensively investigated. The presence of tumor infiltrating NK cells correlates directly with the prognosis and they can also kill cancer stem cells, which tend to be chemo and radio-resistant, and the main responsible for relapses.
A critical aspect in solid tumor treatment is the ability of the cells to migrate and home in the tumor. In the case of inflammation, NK cells move from the blood to the site of inflammation following a chemokine gradient, a process that involves chemokine receptors, adhesion molecules, integrins and other factors. The exact mechanism of migration of NK cells in 3D is not known but the balance of chemoattractants, chemorepellents and the ECM itself may be critical. The use of 3D tumor models and NK cells will allow for a better understanding of these mechanisms. Methods to increase NK cell infiltration have been focusing on the chemokine-chemokine receptor axis. For example, a cleavable NRPbody was designed to recruit NK cells to cancer sites, and included mesothelin ScFv, a furing cleavage site and CXCL16 (chemokine C-X-C motif ligand 16), with the goal to bind to cancer cells via the mesothelin ScFv and recruit NK cells to the tumor by cleavage of the CXCL16 by furin. This approach has shown great promise in animal models by reducing their tumor burden (1, 3).
The blocking of inhibitory checkpoints is also a viable strategy in NK cell therapy. TIGIT and are co-inhibitory receptors, whose ligands are upregulated in cancer cells. TIGIT competes with the activating receptor, DNAM-1, for CD155 and CD112, so it is reasonable to expect that inhibition of TIGIT will remove some of the inhibition cues. The combination of tiragolumab (anti-TIGIT) and atezolizumab (anti-PD-L1) in the treatment of PD-L1 high NSCLC patients showed a response of 66% versus 24% for anti-PD-L1 alone and has already been approved as first line therapy for those patients. Two new inhibitory checkpoints of interest deserve attention: CIS (cytokine-induced STAT inhibitor) and SIRPα (signal regulatory protein alpha).
Challenges on NK Cell Expansion
NK cells have their own specific challenges, such as loss of viability and activity upon freeze/thawing, a natural short period of persistence, the need to be fully activated to be functional, typically they represent a small % of the initial population and dosages range from 5x106 to 1.2x109 NK cells per Kg of patient. The culture conditions ex vivo can have a profound impact on these characteristics (1, 3, 6).
The use of NK cells derived from heterogeneous sources requires that T or B cells are carefully removed, so an initial step of purification/enrichment tends to be desirable. BPS Bioscience offers the NCAM1/CD56 Positive Cell Isolation Kit (BPS Bioscience #78808), which can result in the enrichment of CD56+ cells to a purity level of 97% (Figure 3).
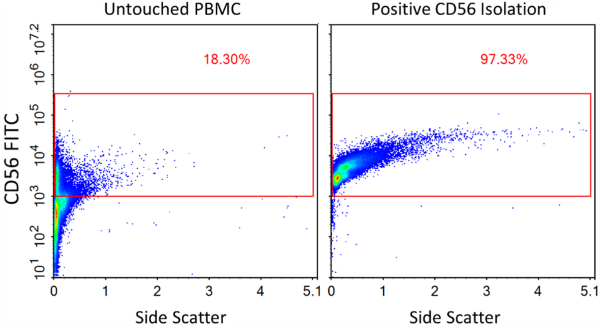
Figure 3: Enrichment of CD56+ cells from PBMCs using NCAM1/CD56 Positive Cell Isolation Kit (BPS Bioscience #78808). Flow cytometry was performed before and after NCAM1/CD56 cell isolation using Anti-NCAM1 Antibody, FITC-Labeled (BPS Bioscience #101865). “Untouched PBMCs” represent the starting PBMC cells while “Positive Selection” represents the population after magnetic separation.
NK cells can be expanded ex vivo by the addition of cytokines and feeder cells. The use of cytokines alone provides a short-term activation but supports only moderate expansion. The use of IL-2 alone even at 1000 U/ml results only in a few cell divisions while pushing cytotoxicity. IL-21 on the other hand can increase ADCC, cytotoxicity and IFNγ production, but cells end up with low viability and in proliferation arrest and apoptosis (6). A combination of feeder cells with cytokines can result in more than 1000-fold expansion over a two-week period of highly cytotoxic cells. Feeder cells can be PBMC or genetically modified K562 cells, where membrane bound IL-15, IL-21, 4-1BBL or OX40L was introduced. With these aspects in mind, we have developed an NK Cell Expansion Kit (BPS Bioscience #78927), composed of Growth-Arrested NK Feeder Cells (K562-modified cells), NK Cell Basal Medium and IL-2. This kit combines all the materials required, can result in high fold expansion (Figure 4). It comes in a convenient format to provide researchers with cytotoxic (Figure 5) NK cells without the need to create their own optimized K562 cell line. We also provide Expanded Human Peripheral Blood NK Cells, Frozen (BPS Bioscience #78798) that can be used directly in assays, such as control in ADCC and cytotoxicity experiments.
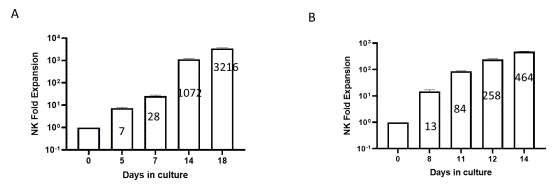
Figure 4: Human NK cell ex vivo fold expansion at different time points during culture. Human PBMCs (A) and isolated NK cells (B) were expanded ex vivo for 14/18 days. Expanded cells were collected on the indicated days and stained with anti-NCAM1 Antibody, PE-Labeled and Anti-CD3 Antibody, FITC-Labeled and analyzed by flow cytometry. The number of CD3-CD56+ cells was determined, and fold expansion was calculated in relation to CD3-CD56+ cell number on day 0.
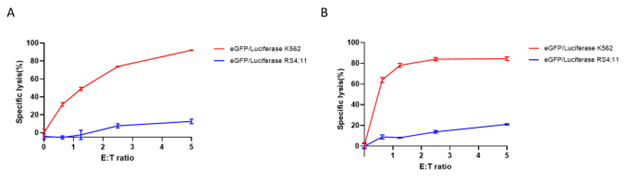
Figure 5: Luciferase activity-based cytotoxicity profile of expanded NK cells. Human PBMCs (A) and isolated NK cells (B) were expanded ex vivo for 14 and 21 days, respectively. Expanded NK cells were co-cultured with eGFP/Luciferase K562 target cells (BPS Bioscience #78911) (red) and eGFP/Luciferase RS4;11 control cells (BPS Bioscience #78926) (blue) at different Effector: Target (E:T) cell ratios, at 37°C for 4 hours. Luciferase activity was detected using ONE-Step™ Luciferase (BPS Bioscience #60690). Cytotoxicity potency (specific lysis) correlates with a decrease in luciferase signal in target cells. Luminescence results were used to generate killing curves.
The use of feeder cells for the production NK cell destined to be used in cell therapy requires that these feeders are irradiated and a careful assessment of the contaminant population at the end is performed. Safety concerns can be minimized for instance by the introduction of a suicide gene into the feeder cells.
The use of iPSC to generate NK cells alleviated some of the challenges of NK cell production, by allowing large doses of homogeneous to be generated. However, methods to differentiate them are challenging and still require feeder cells, such as OP9 stromal cells. Their use is still limited but predicted to increase as differentiation methods become more refined and less time consuming.
Conclusions
NK cells are crucial in cancer immunosurveillance and are indeed natural killers. However, as with T cells, the TME can suppress them and tune them to inhibit other immune cells. The key focus is on the enhancement of their function, generation and expansion. Methods to overcome these technical problems are constantly being optimized, taking NK to the center stage in cancer therapy. BPS Bioscience offers a range of products, from products to generate NK cells, to characterization tools and target cells lines. These include PBMCs, a CD56+ cell isolation kit, cytokines, an expansion kit able to generate high number of active cells, a number of CAR-related tools that can be utilized also in NK cells, target cells lines with reporters for easy assay readouts, and assays kits and recombinant proteins that can be used to identify new molecules that may be critical to potentiate NK cell action. Large steps have been made towards the production and use of NK cells in cell therapy, but it is clear further developments are required to fully take advantage of their enormous potential, particularly for solid tumor treatment.
References
(1) Du N., et al., 2012 Cancers (Basel) 13(16): 4129.
(2) Liu S., et al., 2021 Journal of Hematology & Oncology 14: 7.
(3) Hijjatipour T., et al., 2023 Human Cell. 36 (6): 1843-1864.
(4) Santara S., et al., 2023 Nature 616(7956): 348-356.
│Related Products
│Related Services
- Immunotherapy Biochemical Screening Services
- Immunotherapy Cell-Based Screening Services
- Custom CAR-T Cell Development