Mechanism of JAK-STAT Signaling in Cancer
Understanding cell to cell communication through cytokines and other secreted factors is at the center of daily life for countless pharmaceutical researchers. This is because cytokine-based communication is central to normal development and is often a dysfunctional step in cancer and autoimmune diseases (1, 2). It is also of increasing importance because cytokine release syndrome is a leading cause of toxicology in both bispecific antibody and CAR T-based therapies (3). Much of the cutting-edge research in immunotherapy requires the tireless work of scientists dedicated to studying cellular signaling.
Cytokines, interferons, and growth factors regulate gene expression through the Janus kinase (JAK)-signal transducer and activator of transcription (STAT) pathways. This provides cellular communication essential for normal development of the immune system. Dysregulation of these pathways is associated with myeloproliferative diseases, immune disorders, and several cancers, making members of the JAK-STAT pathways popular targets for therapeutic development (1, 2).
JAK-STAT Pathway
JAK-STAT pathways follow classical signaling models where ligand binds to the extracellular side of a transmembrane receptor and triggers a cascade of phosphorylation reactions (4) (Figure 1). These reactions include phosphorylation of the JAK kinases, the receptor cytoplasmic tail, and STAT transcription factors. STAT phosphorylation leads to nuclear localization, subsequent DNA binding, and gene regulation. Fine control of expression comes from combining a host of ligands and several receptors with four possible JAK proteins (JAK1, JAK2, JAK3 and TYK2), seven STAT family members (STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b, and STAT6), suppressor of cytokine signaling proteins (SOCS), and multiple STAT-dependent operons (5). Control of SOCS expression through STAT-dependent operons leads to a final level of negative feedback inhibition.
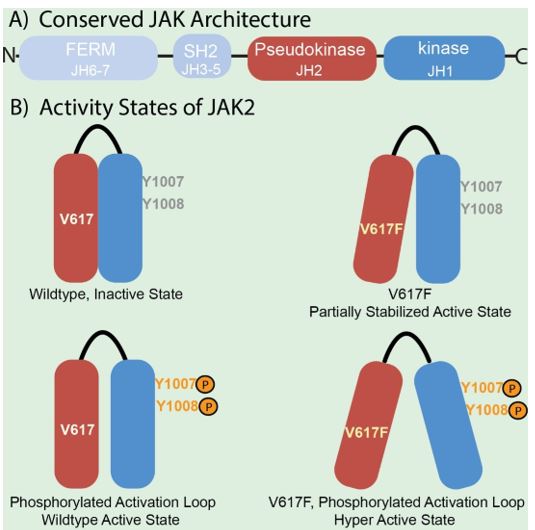
JAK enzymes share a common domain structure (Figure 2) consisting of seven JAK-homology (JH) domains (1, 2). These include the kinase domain (JH1), the inhibitory pseudokinase domain (JH2), the SH2 receptor interaction domains (JH3-5) and the FERM domain (JH6-7). In the simplest model, ligand-receptor binding results in conformational changes in receptor-JAK complexes, allowing phosphorylation of specific tyrosine residues in JH1(6). This promotes additional conformational switches in the JAK proteins that lead to receptor phosphorylation and creating binding sites for STAT proteins.
Multiple mechanisms can contribute to dysregulation of JAK-STAT signaling(1, 2). Aberrant cytokine production, mutations of receptors, and rearrangements and mutations in both JAK and STAT proteins are suspected to cause disease. Gain of function mutations in JAK1, JAK2, and JAK3 are of particular importance. JAK1 and JAK3 mutations are associated with various forms of leukemia and lymphoma. Activating mutations of JAK2 are linked to numerous blood cancers and might be related to STAT1 disfunction in lung, prostate, skin, and breast cancer.
JAK and Disease
The linkage between disease and regulation of JAK activity by the pseudokinase domain is clear. Particularly telling is the overabundance of disease-associated mutations in JH2 domains(1). Genomic studies combined with epidemiology have brought this relationship to light; however, the underlying mechanisms are complex and not completely understood. Releasing the full potential of JAK proteins as drug targets requires detailed biochemical and structural studies of wildtype and mutated forms of the enzymes.
The V617F mutation of the JAK2 JH2 domain is a particularly illuminating example. V617F is a causative agent in myeloproliferative neoplasm patients (7) and linked to overall mortality in several cancers (8). Biochemical domain mapping studies using recombinant JAK2 constructs and varying peptide substrates demonstrate the JH2 domain influences both kinase activity and substrate specificity (9). The V617F mutation increases kinase activity to a level between the inactive and fully active states without effecting substrate-dependent catalytic efficiency. Phosphorylation of tyrosines 1007 and 1008 of the kinase domain activation loop (Figure 2) is necessary for activation of wildtype JAK2 (10, 11). The V617F substitution acts cooperatively with phosphorylation of the activation loop to result in a hyperactive enzyme (6, 9).
The connection between JAK-STAT signaling and disease is attractive to pharmaceutical researchers. Ruxolitinib, an inhibitor of JAK1 and JAK2, was the first JAK inhibitor approved by the FDA (2). It is currently used as a treatment for myelofibrosis and is being investigated for various autoimmune diseases. Tofacitinib was developed as a selective JAK3 inhibitor and is approved for treatment of several forms of arthritis and ulcerative colitis (5). It also inhibits JAK1 and JAK2 in order of decreasing potency (10). Unfortunately, there are known toxicities associated with Tofacitinib, including a newly issued warning from the FDA related to the risk of blood clotting (12). Overcoming toxicity and finding new indications drive a significant amount of research toward finding new JAK inhibitors.
BPS Bioscience understands the drive required for pharmaceutical research. We are dedicated to producing the biochemical and cell-based tools required for advancing drug discovery and for simplifying the work of researchers. Our portfolio includes recombinant JAK proteins for enzymatic and inhibition studies and convenient assay kits designed for use in drug screening. We also offer recombinant cell lines with reporter constructs that are responsive to JAK-STAT pathways and allow for compound testing in a cellular environment. Please contact us to find out more about how we can help you meet your research goals.
References
- H. M. Hammaren, A. T. Virtanen, J. Raivola, O. Silvennoinen, The regulation of JAKs in cytokine signaling and its breakdown in disease. Cytokine 118, 48-63 (2019).
- J. J. O'Shea et al., The JAK-STAT pathway: impact on human disease and therapeutic intervention. Annu Rev Med 66, 311-328 (2015).
- J. N. Brudno, J. N. Kochenderfer, Toxicities of chimeric antigen receptor T cells: recognition and management. Blood 127, 3321-3330 (2016).
- R. Morris, N. J. Kershaw, J. J. Babon, The molecular details of cytokine signaling via the JAK/STAT pathway. Protein Sci 27, 1984-2009 (2018).
- G. A. Durham, J. J. L. Williams, M. T. Nasim, T. M. Palmer, Targeting SOCS Proteins to Control JAK-STAT Signalling in Disease. Trends Pharmacol Sci 40, 298-308 (2019).
- Y. Shan et al., Molecular basis for pseudokinase-dependent autoinhibition of JAK2 tyrosine kinase. Nat Struct Mol Biol 21, 579-584 (2014).
- J. L. Spivak, Narrative review: Thrombocytosis, polycythemia vera, and JAK2 mutations: The phenotypic mimicry of chronic myeloproliferation. Ann Intern Med 152, 300-306 (2010).
- C. Nielsen, H. S. Birgens, B. G. Nordestgaard, L. Kjaer, S. E. Bojesen, The JAK2 V617F somatic mutation, mortality and cancer risk in the general population. Haematologica 96, 450-453 (2011).
- A. Sanz et al., Analysis of Jak2 catalytic function by peptide microarrays: the role of the JH2 domain and V617F mutation. PLoS One 6, e18522 (2011).
- J. Feng et al., Activation of Jak2 catalytic activity requires phosphorylation of Y1007 in the kinase activation loop. Mol Cell Biol 17, 2497-2501 (1997).
- K. Kundrapu, L. Colenberg, R. J. Duhe, Activation loop tyrosines allow the JAK2(V617F) mutant to attain hyperactivation. Cell Biochem Biophys 52, 103-112 (2008).
- FDA, Xeljanz, Xeljanz XR (tofacitinib): Drug Safety Communication - Due to an Increased Risk of Blood Clots and Death with Higher Dose.
Figure 1
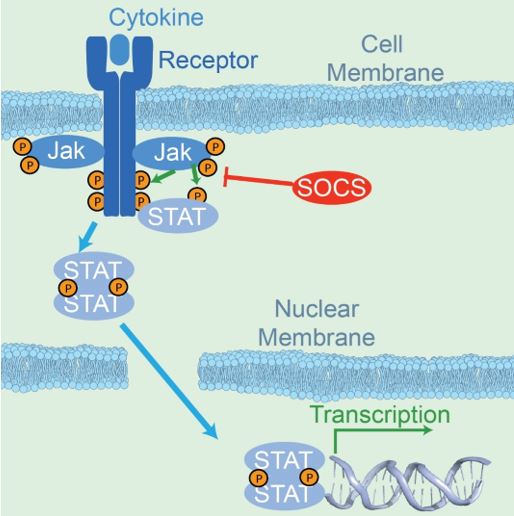
Figure 2
Cell Lines
- GAS Reporter (Luc) – HeLa Cell Line (IFNγ/JAK/STAT1 Pathway)
- ISRE Reporter – HEK293 Recombinant Cell Line (JAK pathway)
Assay Kits
Proteins (WT)
- Jak1, GST-tag
- Jak2 (JH1 domain), His-tag
- Jak2 (JH1, JH2 domain), GST-Tag, Avi-Tag, Biotin (Dephosphorylated)
- Jak2 (JH1, JH2 domain), His-GST-tags
- JAK2 (JH2 Domain), His-Avi-Tag, Biotin-Labeled
Proteins (Mutant)
- Jak2 (JH2 Domain) (E596A, V617F, W659A, W777A, F794H), His-Tag
- Jak2 (JH2 Domain) (V617F, W659A, W777A, F794H), His-Tag
- Jak2 (JH2 Domain) (V617F, W659A, W777A, F794H), His-Avi-Tag, Biotin-Labeled
- Jak2 (JH2 Domain) (W659A, W777A,F794H), His-Tag
- Jak2 (V617F) (JH1, JH2 Domain), GST-Tag
- Jak2 (V617F) (JH1, JH2 Domain), GST-Tag, Avi-Tag, Biotin-Labeled (Dephosphorylated)
- Jak2 (V617F) (JH2 Domain), His-Avi-Tag, Biotin-Labeled
- Jak2 (V617F), FLAG-Avi-Tag, Biotin-Labeled (Desphosphorylated)
- Jak2 (V617F, W659A, W777A, F794H) (JH2 Domain), His-Avi-Tag, Biotin
- Jak2 (W659A, W777A, F794H) (JH2 Domain), His-Avi-Tag, Biotin
- Jak3, His-tag