Claudin-18: A Therapeutic Target in Oncology
Claudins, a family of small tetraspan proteins expressed in a tissue or cell-specific manner, are major components of the multimeric protein architecture of tight junctions, which prevent ions or small molecules from passing freely between epithelial or endothelial cells. These proteins contain four transmembrane domains mainly responsible for interacting side-to-side with neighboring proteins. Two extracellular loops regulate paracellular charge selectivity and form very tight complexes with proteins present on the surface of other cells (trans interaction, or head-to-head interaction), whereas the cytosolic C-terminal tail contains various sites amenable to post-translational modifications, and a protein scaffolding PDZ domain.
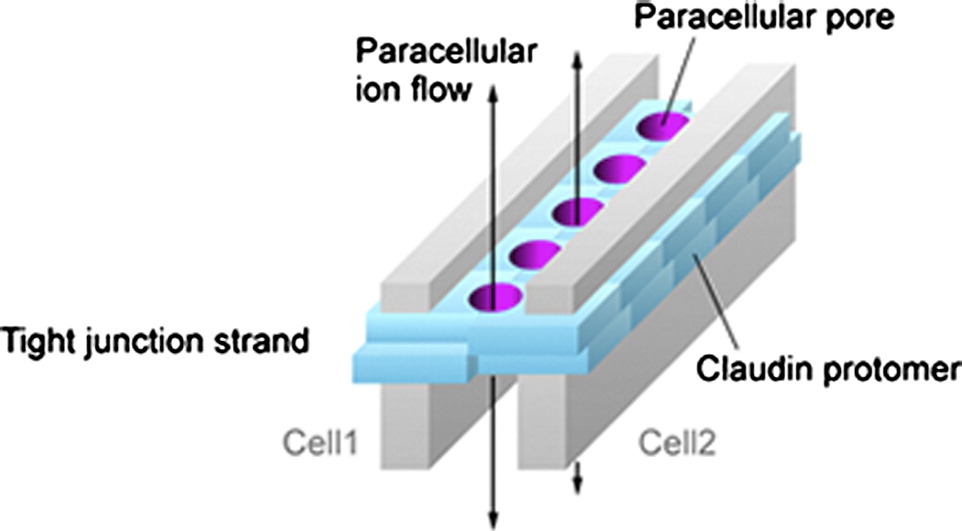
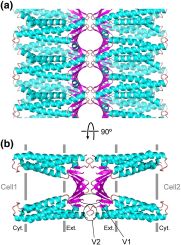
Illustration: Suzuki et al. Journal of Molecular Biology (2015) 427: 291-297; https://doi.org/10.1016/j.jmb.2014.10.020; CC BY 3.0 unported
Claudin-18, and more specifically isoform claudin-18.2, has emerged as a promising therapeutic target in gastric, esophageal, and pancreatic cancer.
Genomics
The CLDN18 gene, located on chromosome region 3q22.3, encodes two isoforms of 261 amino acids (NM_016369.3 and NM_001002026.2 for isoforms 1 and 2, respectively). Claudin-18 is one of the few members in the family to exhibit strict restriction to a cell lineage, a characteristic which is conserved among mammals. Thus, claudin-18.1 is restricted to the alveolar epithelial cells of the lung, whereas claudin-18.2 is exclusively expressed in the differentiated cells of the stomach mucosa.
CLDN18 displays high sequence homology across species, including within the promoter regions (1). The human CLDN18 gene contains 6 exons and 5 introns, with two promoters P1 and P2, each controlling its own first exon. Alternative use of these promoters leads to the production of lung and stomach-specific transcripts 18.1 and 18.2, respectively. In addition, the claudin-18 transcript contains an alternative 12 base-pair insertion containing a stop codon TGA toward the 5’ end of exon 4, which may produce a truncated splice variant, likely resulting in accelerated mRNA decay.
Differences in key amino acids in the first extracellular loop of each isoform confer specific function. For example, claudin-18.2 has a selective sealing function against sodium and protons but not chloride (2).
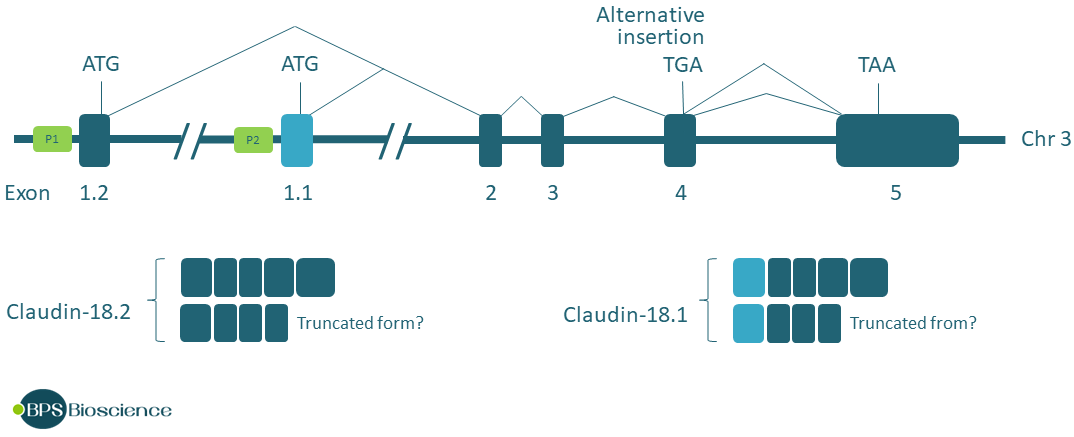
Illustration based on the genomic structure of CLDN18 (3).
The downstream promoter P2 contains binding sites for transcription factor T/EBP/NKX2.1, which is involved in lung embryonic development and becomes restricted to alveolar type II (AT2) and Clara cells. T/EBP/NKX2.1 is responsible for the selective expression of claudin-18.1 in the lung alveolar epithelium and, interestingly, does not induce any other claudin gene (3). The upstream promoter P1, used to produce claudin-18.2, is regulated by transcription factor cyclic AMP-responsive element binding protein (CREB).
Claudin-18 pattern of restricted expression is what makes it so attractive as a cancer target since it will decrease the potential for on-target off-site effects.
Function
Tight junction claudins regulate the permeability of the intercellular space and the circulation of water, specific ions, and macromolecules. In addition, they participate in cell-cell adhesion, contributing to the maintenance of a tight epithelium or endothelium. Claudins not only interact with other proteins present on adjacent cells, but they also associate and cooperate with proteins of the cytoskeleton and with cell signaling proteins through their cytoplasmic PDZ domain, acting as signaling hubs. Claudin-18 typically eliminates the intercellular space by tightening calcium-independent cell-cell adhesion. CLDN18.2, expressed in the epithelial cells of the gastric mucosa, controls paracellular permeability to H+ and Na+.
From the two models of claudin-18-deficient mice that have been generated, it was observed that claudin-18.1 is important for postnatal lung development and for the maintenance of cell architecture (2). Morphologic abnormalities indicated that claudin-18.1 regulates alveolarization and suggested a possible role in pulmonary fibrosis or chronic obstructive pulmonary disorder (COPD), in which alveolar morphology is disrupted. The transgenic mice displayed a surprisingly mild phenotype with respect to barrier functions, even in response to injury, showing almost normal fluid balance. Beyond maintaining the selective permeability of the paracellular barrier, claudin-18 appears to control cell phenotype and inflammation, as suggested by the observation that plasma levels for the pro-inflammatory cytokine IL-1β are significantly increased in claudin-18 deficient mice (2).
Interestingly, the lungs were unexpectedly large in knock out mice, which uncovered a role for claudin-18.1 in the regulation of type 2 alveolar epithelial (AT2) cell proliferation, at least in part driven by translocation to the nucleus of transcriptional regulator Yes-associated protein (YAP). This has important pathological implications, since CLDN18-deficent mice also exhibited an increased frequency of lung adenocarcinoma compared to normal mice (4). The stomach and kidneys of the knock-out mice were also unexpectedly enlarged, although this was not observed in mice in which only claudin-18.2 was targeted possibly due to rescuing effects. Claudin-18 knockout mice eventually develop carcinomas in the lung and stomach.
Thus, the normal function of claudin-18 appears to be tumor suppressive by regulating cellular homeostasis and also controlling cell adhesion and proliferation. It has been proposed that alteration of the structure of tight junctions in epithelia has consequences on morphology, cell migration, and therefore metastasis.
Genetic Alteration of CLDN18
CLDN mutations are very rare in cancer tissue, and no driver mutation has been found for this gene family. On the other hand, it has been observed that CLDN18 gene amplification occurs at a relatively high frequency in Squamous Cell Carcinomas, a histological subtype of carcinoma: over 15% of esophageal squamous cell carcinoma samples and over 10% of lung squamous cell carcinoma samples show CLDN18 amplification (Figure 3).
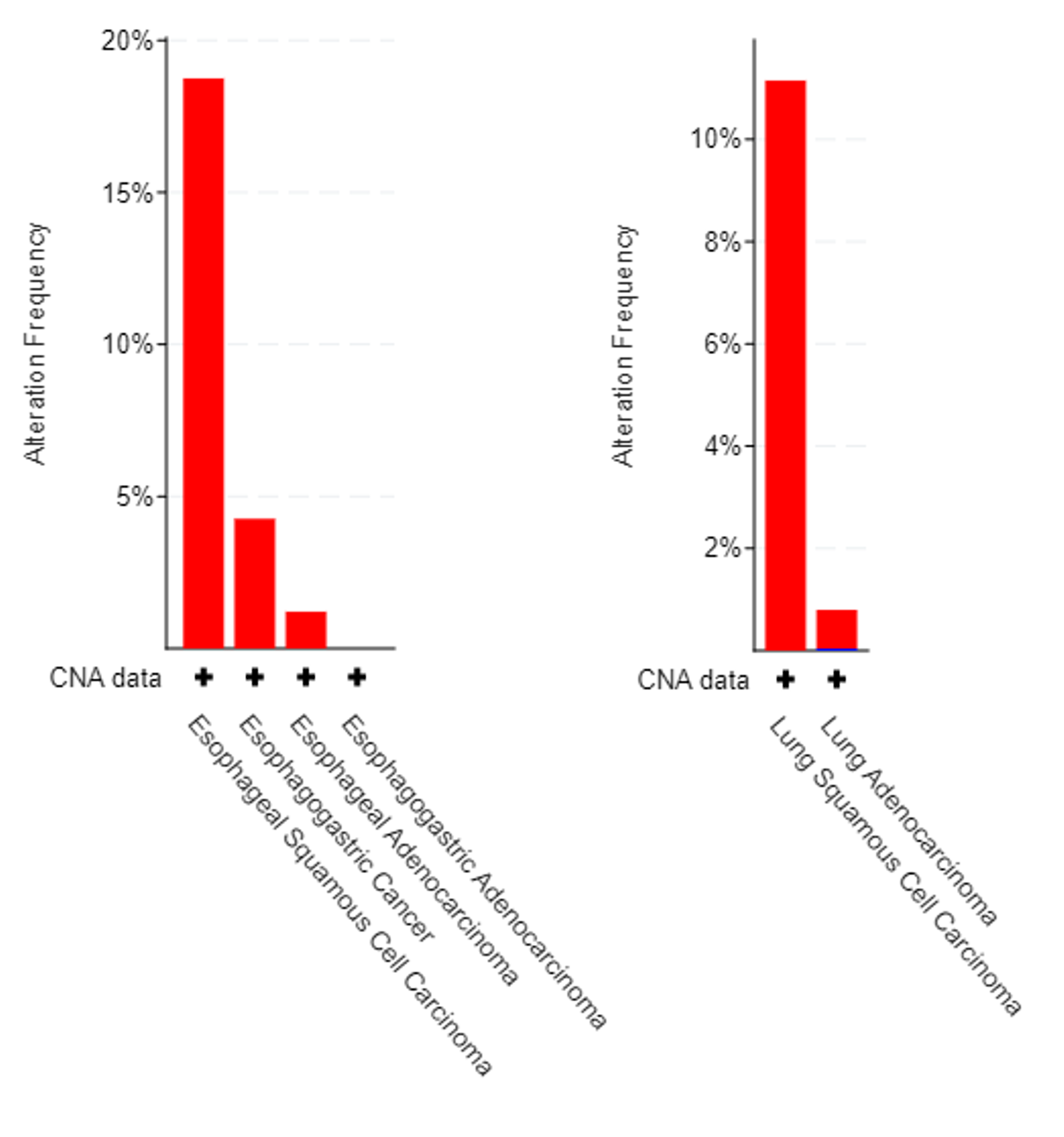
CLDN18 gene amplification in esophageal cancer (left) and in lung cancer (right). Esophageal cancer data correspond to 1191 tumor samples from 503 patients in the 4 studies containing CNA data. Lung cancer data correspond to 9788 tumor samples from 7970 patients in the 10 studies of the dataset containing CNA data. Data was analyzed from the Cancer Genome Atlas https://doi.org/10.1158/2159-8290.CD-12-0095.
In their molecular characterization of gastric adenocarcinoma, the Cancer Genome Atlas Research Network revealed a CLDN18–ARHGAP26 translocation often found in 15% of the genomically stable group (one of four subtypes of gastric cancer). This occurs through the genomic rearrangement of CLDN18 after exon 5 and fusion to exon 2, 10 or 12 of ARHGAP26 (5). Although the fusion occurs downstream of CLDN18 stop codon, a mature mRNA transcript is produced. Use of a cryptic splice site within CLDN18 exon 5 yields an in-frame fusion protein predicted to contain the transmembrane and C-terminus domains of CLDN18. The C-terminus domain is fused to the GAP domain of ARHGAP26, and may affect the activity of small protein RhoA, which regulates cell adhesion and motility, potentially contributing to an invasive phenotype. Patients with this type of gene fusion have worse survival outcomes and show increased resistance to chemotherapy compared to patients who do not have the fusion.
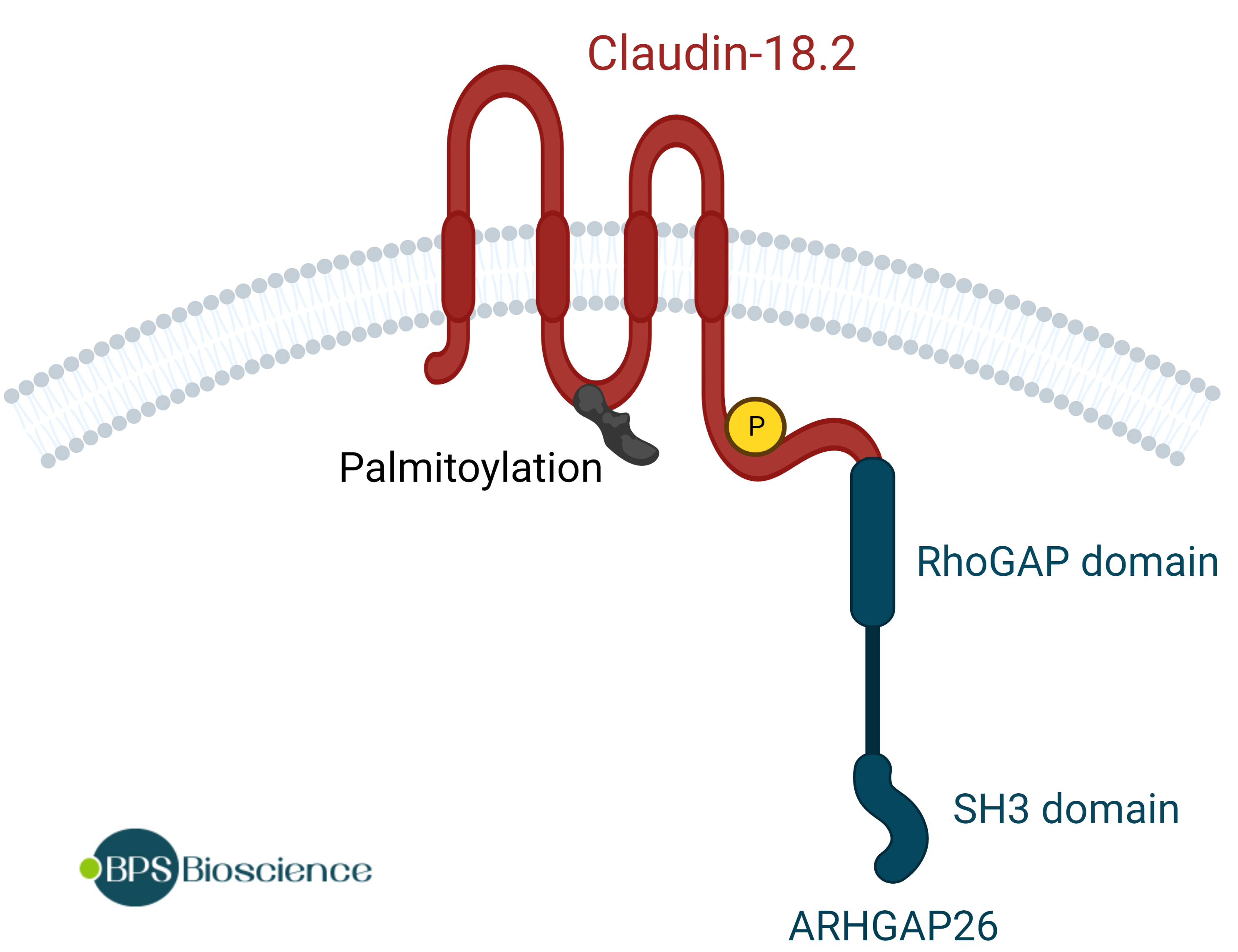
Illustration of the new protein generated by the fusion between claudin-18.2 exon 5 and ARHGAP26 exon 12.
Deregulation of Expression in Cancer
Aberrant cell signaling due to cancerous transformation may result in altered claudin expression or function through a variety of mechanisms, loosening tight junctions and contributing to alteration of tissue morphology, dedifferentiation, migration, and metastasis. In gastric cancer cells for example, downregulation of claudin-18.2 may contribute to tumor-cell proliferation and chemotaxis.
mRNA
Altered expression of claudin-18 has been observed in several types of cancer at the mRNA level or at the protein level (6, 7), however patterns of mRNA expression do not always correlate with patterns of protein expression observed in human tissue. In addition, tumor tissue heterogeneity means that expression may differ between two regions of the same tumor (8). Nevertheless, the current consensus is that mRNA expression of claudin-18.1 is very low to absent in lung cancer tissue compared to normal tissue in which claudin-18.1 expression is high, consistent with a tumor suppressive function. Expression levels of claudin-18.2 mRNA are approximately 60% lower in gastric/esophageal carcinoma tissue compared to normal tissue, possibly correlating with increased hypermethylation patterns in the genomes of gastric tumor samples. Interestingly, although claudin-18.2 is not normally present in the pancreas, it is found upregulated in pancreatic cancer cells (6).
Protein
At the protein level, strong antibody staining is consistently observed in human tissue specimens obtained from patients with gastric, esophageal, or pancreatic cancer. Depending on the study, claudin-18.2 was robustly expressed in 20% to 80% of all gastric and pancreatic cancer tissue samples (8-11). All studies found that claudin-18.2 expression levels in the metastatic lesions are consistent with expression levels in the corresponding primary cancer tissues, which is a clinically significant consideration for targeted therapy since most patients die of metastasis. Interestingly, common gastric cancer cell lines such as 293T, AGS, and BGC-823, may not express claudin-18.2 at all (10).
The aberrant expression of claudin-18.2 in about 50% of pancreatic adenocarcinoma, but not in the normal tissue, creates a unique therapeutic opportunity for this hard-to-treat cancer (11, 12). Indeed, preclinical studies have now shown that anti-claudin-18.2 antibodies inhibit patient-derived pancreatic xenograft tumors in vivo [Zhu 2019].
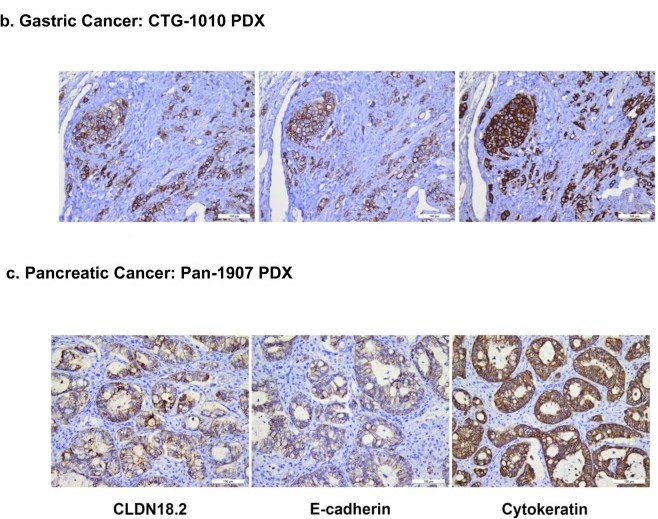
Patient-derived gastric and pancreatic tumors, stained with anti-CLDN18.2 antibody (left), and with antibodies against human cytokeratin (middle) and E-cadherin (right). Reproduced without modification from (8); doi: 10.1038/s41598-019-44874-0. CC BY 4.0
Epithelial-Mesenchymal Transition
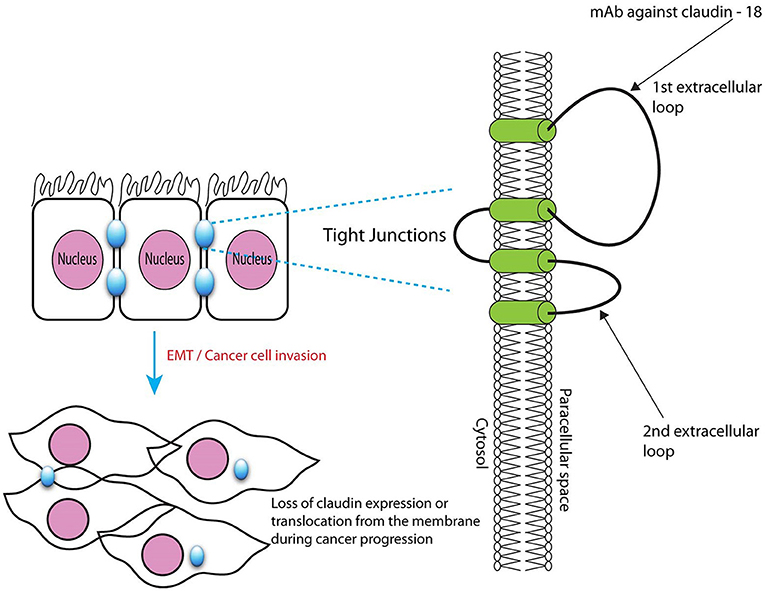
Claudin-18.2 protein expression inversely correlates with the degree of epithelial-mesenchymal transition (EMT) or with stemness. Thus, the protein is absent from the stem cell zone of gastric glands and the expression of claudin-18.2 is low in tumors that arise from these stem cells. Conversely, claudin-18.2 expression is elevated in differentiated gastric cells and decreases when the cells transition into an undifferentiated state or progress through EMT transformation (13).
Since EMT causes a loss in epithelial organization of the tissue and disorganization of cell-cell junctions, claudin-18.2 becomes delocalized. Proper localization of claudin-18.2 in apical tight junctions makes it mostly inaccessible to antibodies, whereas the extracellular domain of the protein becomes accessible in tumor tissue which shows mostly disorganized tumor cells lacking polarity. By itself, this phenomenon contributes greatly to the attraction of claudin-18.2 as a therapeutic target.
Biomarker and Molecular Imaging
Enhanced antibody staining for claudin-18 and high selectivity of expression in gastric or pancreatic adenocarcinomas suggests that the protein could be used as a biomarker for companion diagnostics to select patients most likely to benefit from antibody-based therapeutic strategies. In addition, labeled antibodies may be used in conjunction with positron emission tomography (PET) to delineate tumor boundaries prior to surgery and to guide surgical resection (14).
Therapeutic Opportunities
Owing to its restricted expression in gastric, esophageal, and pancreatic cancer, including metastases, claudin-18.2 is a highly promising therapeutic target to treat these types of cancer. Increased exposure of the protein extracellular domain to antibodies as occurs during tumor progression makes it quite ideal for antibody-based approaches.
Patients with advanced gastroesophageal (GE) adenocarcinomas and pancreatic adenocarcinomas have dismal 5-year survival rates of 5-20% and 6-8%, respectively. The median overall survival is about 3.5 months for patients with advanced pancreatic cancer, and 10 months for patients with GE cancer, therefore new therapeutic modalities are urgently required. Approximately 30% of these tumors overexpress claudin-18.2, offering new options for these patients who currently don’t have many.
Ideally, a therapeutic agent binds preferably to the cancer cells and not to normal tissue. A target that is present or accessible only in cancer cells, such as claudin-18.2, is attractive because on-target side effects are reduced, providing a broader therapeutic window. However, isoform 18.1 differs from 18.2 by only 7 amino acids (located in the first extracellular loop ECL1), which is an obstacle to the development of highly selective antibodies against claudin-18.2. Nevertheless, various therapeutic strategies are now under development to take advantage of the differential accessibility of claudin-18.2 in cancer cells. These strategies include monoclonal antibodies, claudin-directed chimeric antigen receptors (CAR) for cell adoptive transfer, an anti-claudin-18.2/PD-L1 bi-specific antibody, anti-claudin/anti-CD3 bi-specific T cell engager and a tetravalent engager, and antibody drug conjugates (7, 15).
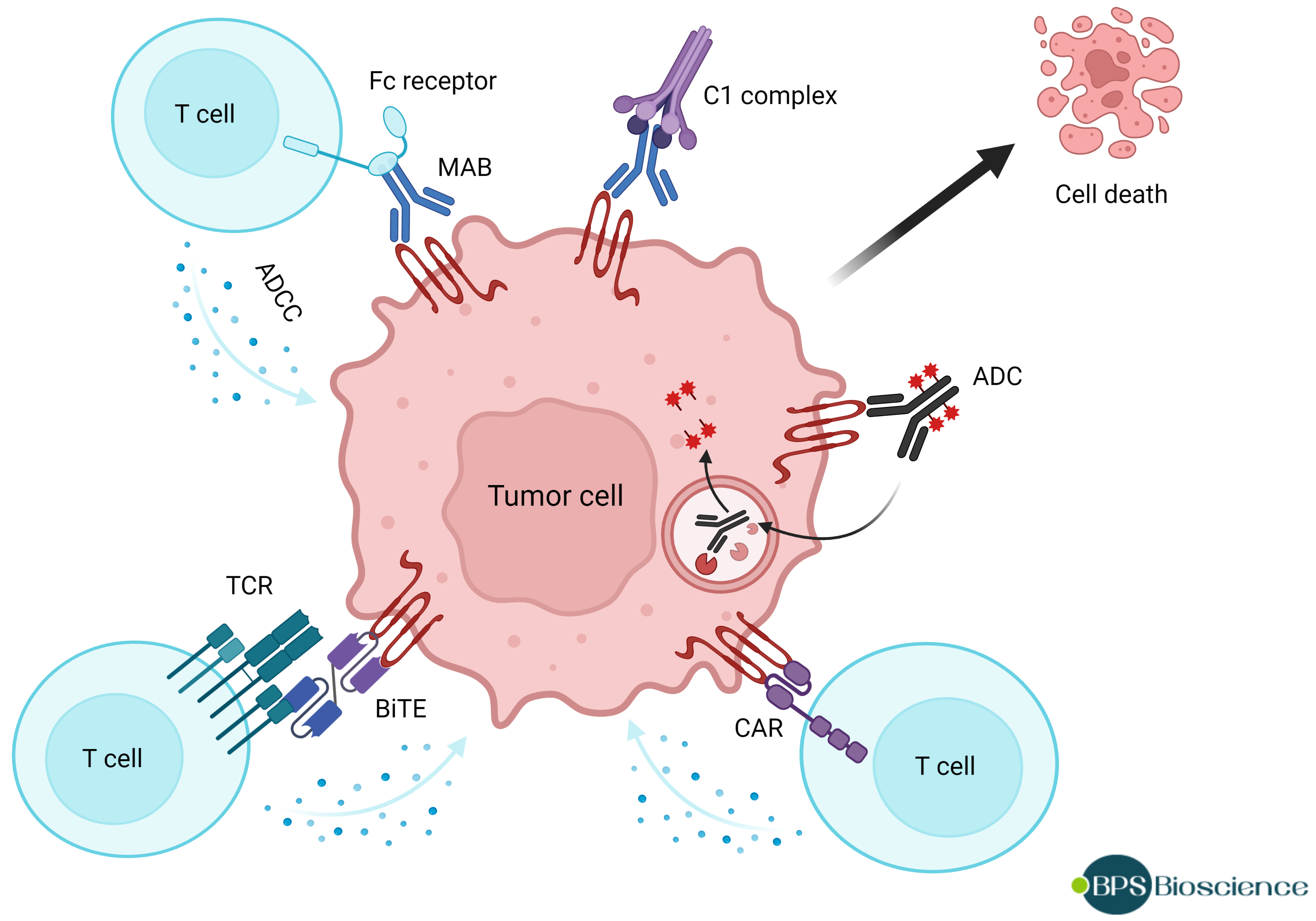
Illustration of various therapeutic strategies targeting claudin-18.2. ADC: antibody-drug conjugate; ADCC: antibody-dependent cellular cytotoxicity; BiTE: bi-specific T cell engager; CAR: chimeric antigen receptor; MAB: monoclonal antibody; TCR: T cell receptor.
1) Monoclonal Antibody Against Claudin-18.2
The first claudin-directed therapeutic antibody to establish proof-of-concept and reach late phase clinical trial was Zolbetuximab, also known as IMAB362 and claudiximab, developed by Ganymed Pharmaceuticals AG for the treatment of claudin-18.2-positive gastric, esophageal, and pancreatic adenocarcinoma. The antibody is a humanized chimeric IgG1 monoclonal antibody against the extracellular loop claudin-18.2 that is highly specific to claudin-18.2 and does not recognize claudin-18.1. IMAB362 demonstrated very good tumor selectivity compared to normal tissue and in preclinical studies it induced cell death in CLDN18.2+ human xenografts.
Mechanistically, IMAB362 mediates cell death by engaging the immune system through antibody-dependent cellular cytotoxicity (ADCC) and complement-dependent cytotoxicity (16). The clinical efficacy demonstrated in the FAST trial for gastric and gastroesophageal junction (GEJ) cancer, which included increased overall survival, validated claudin-18.2 as a therapeutic target and paved the way for use of anti-claudin-18.2 antibodies and for further development.
Efficacy was associated with high claudin-18 expression in >70% of the cells, providing a prognosis marker and cut-off point (13). The combination of the antibody with chemotherapy would be expected to increase ADCC, and indeed the antibody demonstrated a survival benefit in combination with standard chemotherapy compared to chemotherapy alone, in advanced claudin-18.2-positive gastric and gastro-esophageal cancer. The antibody is now under investigation for both gastrointestinal adenocarcinomas and pancreatic tumors.
2) Antibody-Drug Conjugates
The first two antibody drug conjugates (ADC) targeting claudin-18.2 were CPO102, developed by Conjupro Biotherapeutics, and SOT102 from SOTIO and NBE Therapeutics. CPO102 is an anti-claudin-18.2 monoclonal antibody conjugated to monomethyl auristatin E (MMAE) conjugates. MMAE, also known as vedotin, is an antimitotic agent about 100-1000 times more potent than doxorubicin that acts by blocking tubulin polymerization. MMAE is so potent that it can’t be used as a drug by itself, however it is frequently used as payload in ADCs since the antibody restricts the drug distribution in the body, which greatly decreases systemic toxicity.
Drug-conjugated antibodies differ by the specific antibody portion used in the construct, by the number of drug molecules appended, by the chemistry and size of linker, and by the mechanism of activation which intrinsic to the design of the linker. Thus, CPO102 is conjugated to MMAE using a linker that is stable in the plasma and extracellular milieu but is cleaved by cathepsin once the ADC has found its target and entered the tumor cell, after which MMAE is released from the antibody. SOT102, on the other hand, uses sortase-mediated antibody coupling for conjugation in a site-specific manner. A non-cleavable amide/peptide linker is used to attach a derivative of PNU159682, which is a highly potent metabolite of the anthracycline nemorubicin which acts as a DNA topoisomerase II inhibitor. The effector function of the anti-claudin-18.2 antibody has been modified to decrease FcRγ interaction while maintaining FcRn binding.
3) Bi-specific Antibodies
Anti-CD3 bispecific antibodies are constructed from two separate antibodies, one that binds to a target protein on tumor cells and the other that binds to the CD3 subunit of the T cell receptor (TCR) on the surface T cells. Thus, these antibodies are used to redirect immune cells to tumors and induce T cell-mediated cytotoxicity.
In their proof-of-concept study, Zhu et al reported the design of humanized CD3ε x claudin-18.2 bispecific antibody and diabody consisting of a human IgG2 Fc fragment with mutations that reduce Fcγ receptor binding. These antibodies induced similar cytotoxicity in cellular models of gastric and pancreatic cancer. Both constructs inhibited the growth of gastric patient-derived xenograft tumors implanted in immuno-compromised NSG mice, although the diabody had better in vivo efficacy compared to the bispecific antibody (8).
4) CAR T Cells
The first claudin-18-directed autologous CAR-T cell therapy to reach the clinic was CT041, developed by CARsgen Therapeutics, Ltd. CARsgen’s scientists developed a humanized monoclonal antibody specific for the claudin-18.2 isoform and used the single-chain fragment variables (scFv) to construct the CAR, which was transduced into T cells via lentivirus infection. In vivo evaluation in mice bearing tumor xenografts derived from gastric cancer patients showed partial or complete tumor elimination without obvious toxicity, even in the normal gastric tissue (10).
CT041 is in clinical trial both in the USA and in China, where gastric cancer accounts for over 40% of the world total number of cases. Preliminary results in patients with very advanced disease indicated a 60% overall response rate, higher than historical data would predict, which bodes well for the future of anti-claudin-18.2 CAR-T cell therapy.
Conclusion
With promising clinical trial results and various therapeutic approaches in development, the future is bright for claudin-18-directed therapeutics. So far it appears that the side effects of targeting claudin-18 are relatively mild, which will further motivate drug development efforts. Clinical trials in pancreatic cancer are under way, a ray of hope for this difficult to treat cancer.
References
(1) Türeci O, et al. Claudin-18 gene structure, regulation, and expression is evolutionary conserved in mammals. Gene (2011) 481(2): 83-92. PMID: 21571049.
(2) Schlingmann B, et al. Claudins: Gatekeepers of lung epithelial function. Semin Cell Dev Biol. (2015) 42: 47-57. PMID: 25951797.
(3) Niimi T, et al. Claudin-18, a novel downstream target gene for the T/EBP/NKX2.1 homeodomain transcription factor, encodes lung- and stomach-specific isoforms through alternative splicing. Mol Cell Biol. (2001) 21(21): 7380-90. PMID: 11585919.
(4) Zhou B, et al. Claudin-18-mediated YAP activity regulates lung stem and progenitor cell homeostasis and tumorigenesis. J Clin Invest. (2018) 128(3): 970-984. PMID: 29400695.
(5) Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. (2014) 513(7517): 202-9. PMID: 25079317.
(6) Li J. Dysregulated expression of claudins in cancer. Oncol Lett. (2021) 22(3): 641. PMID: 34386063.
(7) Kyuno D, et al. Claudin-18.2 as a therapeutic target in cancers: cumulative findings from basic research and clinical trials. Tissue Barriers. (2022) 10(1): 1967080. PMID: 34486479.
(8) Zhu GF, et al. Targeting CLDN18.2 by CD3 Bispecific and ADC Modalities for the Treatments of Gastric and Pancreatic Cancer. Sci Rep. (2019) 9(1): 8420. PMID: 31182754.
(9) Mitnacht-Kraus R, et al. Preclinical characterization of IMAB362 for the treatment of gastric carcinoma. Abstracts developmental therapeutics (2017) 28(S5): V126; Open Archive.
(10) Jiang H, et al. Claudin18.2-Specific Chimeric Antigen Receptor Engineered T Cells for the Treatment of Gastric Cancer. J Natl Cancer Inst. (2019) 111(4): 409-418. PMID: 30203099.
(11) Tanaka M, et al. Claudin-18 is an early-stage marker of pancreatic carcinogenesis. J Histochem Cytochem. (2011) 59(10): 942-952. PMID: 21832145.
(12) Li J, et al. Analysis of the expression and genetic alteration of CLDN18 in gastric cancer. Aging (Albany NY). (2020) 12(14): 14271-14284. PMID: 32668412.
(13) Sahin U, et al. FAST: a randomised phase II study of zolbetuximab (IMAB362) plus EOX versus EOX alone for first-line treatment of advanced CLDN18.2-positive gastric and gastro-oesophageal adenocarcinoma. Ann Oncol. (2021) 32: 609-619. PMID: 33610734.
(14) Cao W, et al. Claudin18.2 is a novel molecular biomarker for tumor-targeted immunotherapy. Biomark Res. (2022) 10(1): 38. PMID: 35642043.
(15) Zhang J, et al. Evaluation and reflection on claudin 18.2 targeting therapy in advanced gastric cancer. Chin J Cancer Res. (2020) 32(2): 263-270. PMID: 32410803.
(16) Türeci Ӧ, et al. Characterization of zolbetuximab in pancreatic cancer models. Oncoimmunology (2018) 8(1): e1523096. PMID: 30546962.