Setting a Trap for PARP1 and PARP2
Summary
PARP1 and PARP2 operate in DNA damage response (DDR) by sensing single breaks in DNA and by recruiting the repair machinery through poly-ADP ribosylation of histones and other proteins. PARP inhibitors are cytotoxic to cancer cells deficient in repair pathways that are complementary to PARP pathways, and it has been shown that some of these inhibitors act by trapping PARP1/2 onto the DNA, preventing repair and leading to cell death. Thus, DNA trapping is a highly desirable property of PARP inhibitors.
To support the discovery of new PARP inhibitors with trapping ability we have developed an innovative PARPtrap™ assay designed for high-throughput screening of small molecule libraries and specifically identify or compare inhibitors capable of trapping PARP1 and/or PARP2 onto DNA (1).
Introduction
DNA damage, resulting in base mismatches, bulky adducts, cross-links, single strand breaks, or double strand breaks, occur from a variety of exogenous and endogenous mechanisms. To maintain genome integrity in response to DNA damage, mammalian cells employ an array of repair mechanisms. The DNA repair pathway that is initiated depends on the type of damage and is highly context dependent. Repair pathways can also trigger cell cycle blockade, stress responses, and apoptosis.
Poly-ADP Ribose Polymerase (PARP) is a large family of proteins consisting of 17 members, which are key initiators of the DDR (2). Among the PARP family, PARP1 to 3 are specifically involved in damage sensing, in the recruitment and activation of repair proteins at the site of damage, in genome maintenance, and cell death. PARP1 and PARP2 are mainly involved in DNA repair, and both proteins regulate the DDR network. PARP2 also regulates epigenetic, proliferative, and inflammatory processes and is important for spermatogonia, thymus, and adipose tissue development (3, 4). In contrast, PARP1 alters transcription and induces apoptosis when DNA is damaged beyond repair and therefore may play a more prominent role as a therapeutic target.
Since maintaining genomic integrity is critical for an organism, at least 150 different proteins form the intricate DDR network that constantly scans and repairs DNA (5). Indeed, in the absence of proper repair, genomic instability leads to the accumulation of genetic changes that can be directly responsible for the formation of tumors. For example, BRCA1 (breast cancer type 1 susceptibility protein 1) and BRCA2 mutations impair cellular ability to repair double-stranded DNA breaks through homologous recombination (HR), increasing the person’s susceptibility to breast, ovarian, or prostate cancer (3). However, the loss of HR-dependent DNA repair means that these tumor cells rely on other repair pathways for survival, exposing a therapeutic Achille’s heel.
This has been exploited by scientists in the context of synthetic lethality, which is cell death resulting from the simultaneous disruption of two proteins, whereas there is no loss of viability when each protein is impaired individually. Thus, HR deficiency by itself does not kill a cell, but adding PARP inhibitors efficiently targets HR-deficient cancer cells for apoptosis by specifically blocking a complementary DNA repair pathway (6).
Four PARP1/2 inhibitors are currently approved for clinical use in the USA, with two more approved in China and many others making their way through (pre)clinical phases.
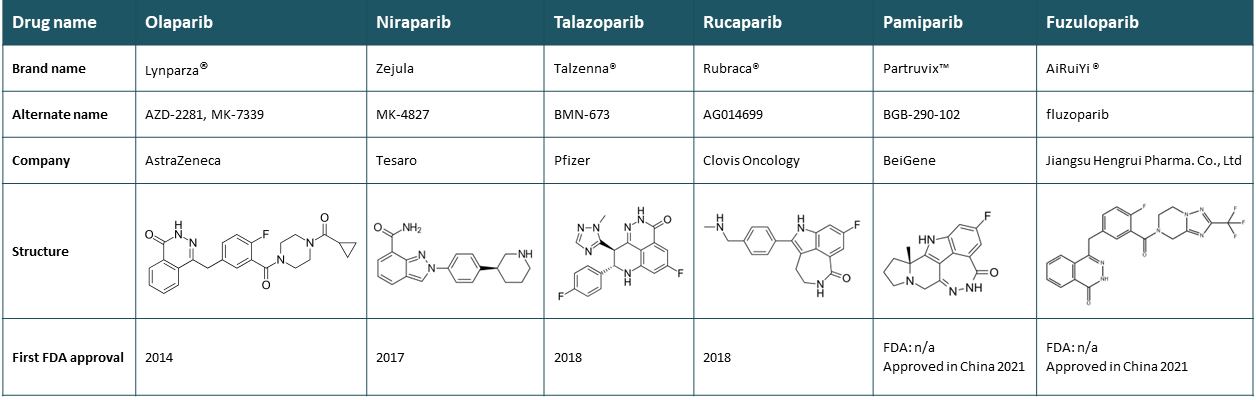
Table 1: PARP inhibitors currently used in the clinic.
New clinical applications of approved inhibitors are still expanding as it is now established that blocking any HR pathway in tumor cells (not limited to BRCA genes) confers “BRCAness” and is lethal to cells in the presence of PARP inhibitors. Indeed, PARP inhibitors also inhibit DNA repair in response to radio- or chemotherapies that induce single-stranded DNA breaks, paving the way for this therapeutic class to treat various tumors in diverse therapeutic combinations (4, 7).
Discovery of DNA trapping as PARP inhibitors mechanism of action
Approximately ten years after the first PARP inhibitor entered clinical development, it was discovered that in addition to blocking the enzymatic activity of PARP1, PARP inhibitors can also lock PARP onto the DNA (8).
What is DNA trapping?
As first responders, PARP1 and PARP2 sense single strand breaks in the DNA and immediately bind to it before adding poly-ADP ribose (PAR) chains to their own protein backbone, to histones, and to other repair proteins in order to recruit and activate them. The PARylated PARP then detaches from the DNA, due to the strong electrostatic charge of the PAR chains, allowing other proteins to initiate the repair process. Removal of PAR chains by PARG (Poly (ADP-ribose) glycohydrolase) recycles PARP to its inactive form and ready to sense DNA damage again.
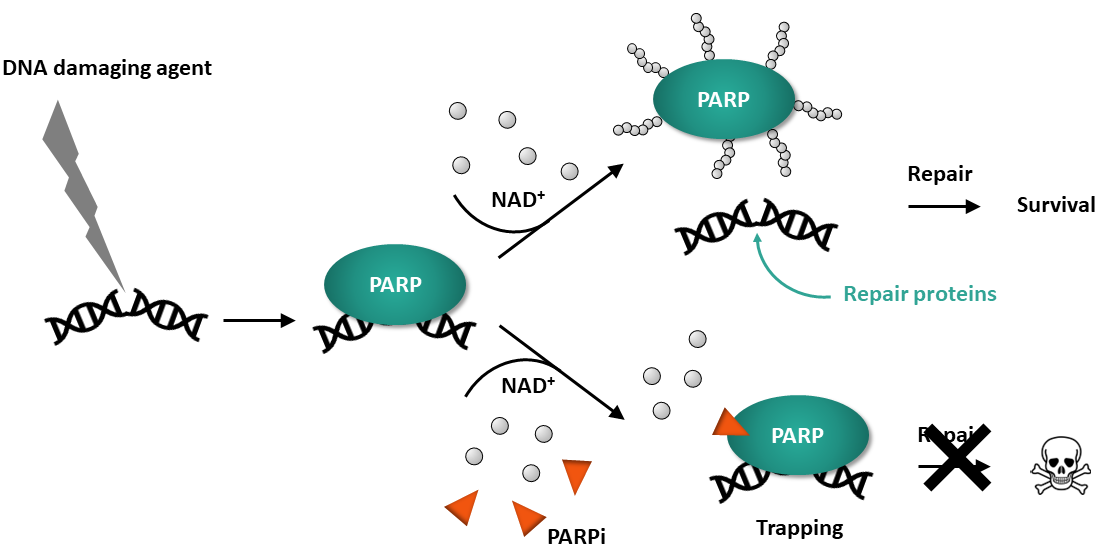
Figure 1: PARP binds to a DNA single-strand break and is activated. Auto-PARylation decreases its affinity for DNA, resulting in dissociation. A PARP inhibitor that prevents release from the DNA is said to “trap” PARP onto DNA.
Currently approved drugs reduce the activity of PARP1 and PARP2 by competing with NAD+ binding and preventing auto-PARylation, causing failure to detach from the DNA. The continuous presence of PARP at the site of damage prevents repair and blocks replication, leading to cell death. Therefore, drugs that trap PARP to the DNA tend to be significantly more cytotoxic than PARP inhibitors with weak trapping ability (9).
Out of four clinical drugs that inhibit PARP1/2, three differ in their potency to trap PARP1 and PARP2 irrespective of their potency to inhibit PARP catalytic activity (4, 8, 9). Indeed, the cytotoxic effects of this class of drugs and their clinical efficacy seem to correlate with how efficiently they trap the PARP protein to the damaged DNA, acting as cellular poison (10).
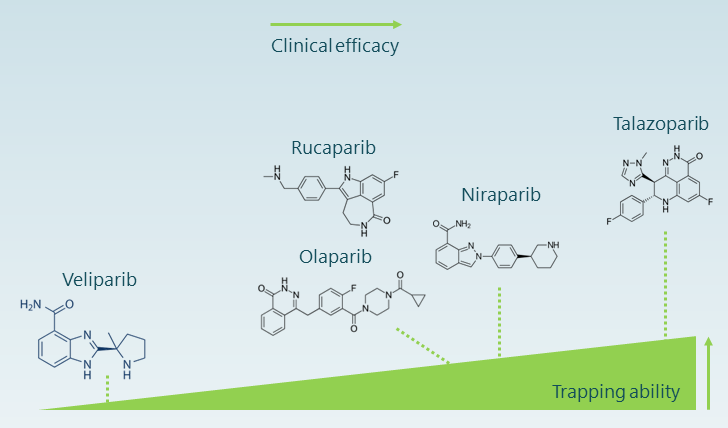
Figure 2: Increasing clinical efficacy of PARP inhibitors in relation to their known ability to trap DNA.
In addition, scientists recently observed that trapping PARP1 to DNA, but not PARP2, results in increased cytotoxicity. Therefore, screening for such inhibitors should include assays that quantify PARP-trapping ability and distinguish an inhibitor selectivity to PARP1 or PARP2 (3, 8).
PARPtrap™ assays specifically assess the ability of a drug to trap PARP onto DNA.
Most commercially available PARP assays measure its enzymatic activity and quantify the PARylation of target proteins, such as histones. In contrast, PARPtrap™ assays measure a compound’s ability to keep PARP1 or PARP2 onto a DNA probe, instead of measuring its effect on PARP enzymatic activity.
This homogeneous, simple assay can be incorporated into high-throughput drug discovery screens or rounds of optimization for molecules that enhance trapping. PARPtrap™ assays allows researchers to efficiently screen their libraries for the most effective inhibitors.
The assay is based on principles of fluorescence polarization and uses fluorescently labeled DNA probes, which, when excited by polarized light, emit fluorescence with a degree of polarization proportional to the rate of molecular rotation. Free DNA probes are very small and rotate fast, therefore they have low fluorescence polarization (FP). When in complex with PARP, molecular rotation is slow due to the large size of the protein, and fluorescence polarization is high. Upon addition of NAD+, the newly PARylated enzymes detach from the probe, reducing FP levels (see Figure 3).
In practice, the user incubates purified PARP1 or PARP2 with a specific fluorescent DNA probe, then adds NAD+ to initiate PARylation. Once PARylated, PARP detaches from the probe due to the high negative charge of the PAR chains. This is the experimental condition in which FP is the lowest since most of the DNA probe will be free.
Pre-incubation of PARP with an inhibitor that blocks PARylation, but not the binding to the DNA, will keep the enzyme trapped to the DNA probe, a condition in which the probe remains engaged in a complex with PARP, resulting in high FP. Thus, the user compares a no-inhibitor control at low FP and a test inhibitor condition at high FP, which means that an increase in the FP signal indicates trapping by the inhibitor.
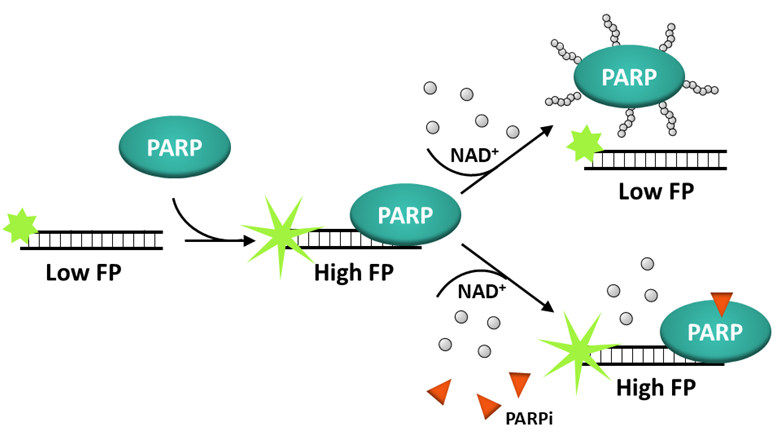
Figure 3: Principle of the PARPtrap™ Assay.
For a more detailed review of the principles of fluorescence polarization and tips on the use and analysis of FP assays, visit our FAQs and Tech Note. Keep in mind that the assay requires a fluorescent plate reader capable of reading fluorescence polarization (capable of polarized light emission and measurement of the difference in fluorescence between horizontal and vertical panes).
Comparison of the potency of known inhibitors in trapping PARP1 and PARP2
Two assays, corresponding to PARP1 and PARP2, were optimized for probe specificity and enzyme kinetics, displaying similar range and sensitivity. Each assay uses enzymatically validated purified PARP1 or PARP2 proteins.
In the experiments shown in Figure 4, PARP1 trapping (left) and PARP2 trapping (right) were measured in the presence of increasing concentrations of Talazoparib, Olaparib, Veliparib, and AZD5305 using the PARPtrap™ Assay Kit for PARP1 (#80584) and the PARPtrap™ Assay Kit for PARP2 (#78296), respectively. “No compound” corresponds to the lowest FP allowed by the assay and “no NAD+” corresponds to the highest FP allowed by the assay. Efficacy, as measured by EC50, is indicated for each drug on the two graphs.
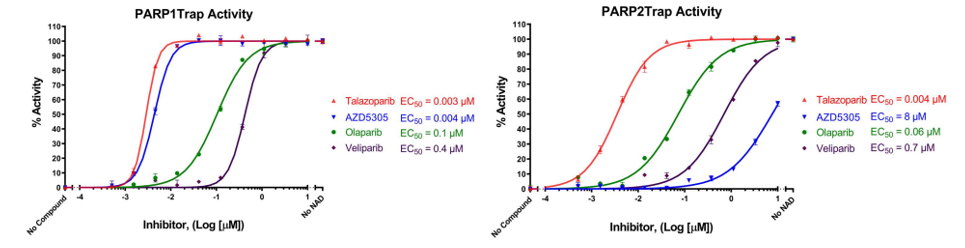
Figure 4: Trapping of PARP1 and PARP2 by PARP inhibitors. PARP trapping on DNA measured in the presence of increasing concentrations of Talazoparib, Olaparib, Veliparib, and AZD5305 (#78318) using the PARPtrap™ Assay Kits, BPS Bioscience, #80584 & #78296. “No compound” corresponds to the “Low FP control” and “no NAD” corresponds to the “High FP control”.
Talazoparib, Olaparib and Veliparib had similar trapping efficacy against PARP1 and PARP2, as measured by their EC50, whereas AZD5305 was 1,000 times more efficient at trapping PARP1 than PARP2, demonstrating selectivity between PARP1 and PARP2. The relative trapping efficacies observed in the assay for Talazoparib, Olaparib and Veliparib were similar to known relative clinical efficacies (8). In these assays, AZD5305 displayed DNA trapping activity toward PARP1 as efficient as Talazoparib.
These results indicate that the assays, by quantifying PARP-trapping ability, can distinguish an inhibitor selectivity to PARP1 or PARP2, providing clues as to which inhibitor may prove more cytotoxic.
Conclusion
BPS Bioscience developed a homogeneous, simple assay that can be incorporated into high-throughput drug discovery screens for molecules that enhance PARP1 or PARP2 trapping on DNA. Thus, PARPtrap™ is a unique assay that enables scientists to efficiently screen their libraries for the most specific and effective inhibitors. In addition, the PARPtrap™ Combo Assay Kit for PARP1 and PARP2 (#78317) compares a molecule’s ability to trap PARP1 versus PARP2 in the same assay, allowing direct comparison of a compound’s potency on each of the PARP isozymes.
Advantages
- Simple procedure
- Only 3 hours start-to-finish
- No-washes
- Detects differences in compound potency against PARP1 and PARP2
- 96-well or 384-well
References
(1) Zientara-Rytter K, et al. 2023 Cancer Research 83(7_Supplement): 6111.
(2) Ummarino S, et al. 2021 Genes 12(3): 446.
(3) Ali SO, et al. 2016 Am. J. Cancer Res. 6(9): 1842-1863.
(4) Rose M, et al. 2020 Front. Cell Dev. Biol. 8: 564601.
(5) Wood RD, et al. 2001 Science 291: 1284-1289.
(6) Rudolph J, et al. 2022 PNAS USA, 119(11): e2121979119.
(7) Lord CJ, Ashworth A. 2016 Nat. Rev. Cancer 16(2): 110-120.
(8) Murai J, et al. 2012 Cancer Res. 72: 5588–99.
(9) Krastev DB, et al. 2021 Cancer Res. 81(22): 5605-5607.
(10) Pommier Y, et al. 2016 Sci. Transl. Med. 8(368): 368er7.