AAV and Gene Therapy
Introduction
![]() Adeno-Associated Viruses (AAV) have become a prominent tool for gene delivery and have been used with success to treat diseases where a gene is missing, requires editing or silencing, and to increase longevity. The ability to manipulate its capsids, for instance, has advanced the gene therapy (GT) field. Here we will discuss AAV biology, its applications in gene therapy, and current challenges.
Adeno-Associated Viruses (AAV) have become a prominent tool for gene delivery and have been used with success to treat diseases where a gene is missing, requires editing or silencing, and to increase longevity. The ability to manipulate its capsids, for instance, has advanced the gene therapy (GT) field. Here we will discuss AAV biology, its applications in gene therapy, and current challenges.
AAV Biology
AAV were initially found as contaminants in adenovirus preparations, a finding that gave them their name. They belong to the parvoviridae family, and require co-infection with a helper DNA virus, such as adenoviruses, to be able to replicate. They are small non-enveloped viruses with a single stranded 4.8 kb DNA genome that contains three genes, which can generate nine proteins, flanked by ITRs (inverted terminal repeats). The Rep (replication) gene encodes four proteins involved in viral replication and packaging. Cap (capsid) generates the capsid proteins, VP1/2/3. These form the typical icosahedral structure of the virus and are required for viral protection, binding, and internalization. The third gene, aap (assembly), encodes AAP (assembly activating protein), which seems to be essential for nuclear localization of VP proteins and capsid assembly in some serotypes, but seems totally redundant in others (1, 2). The only elements strictly necessary for DNA replication and packaging are the ITRs, which allow the remainder of the genome to be replaced with DNA of interest (2).
AAVs interact with carbohydrates on the surface of host cells, such as sialic acid, galactose and heparin sulfate. Following this primary contact, which may induce a capsid conformational change, there is an interaction via receptors. The efficiency of transduction of a certain serotype in a specific cell type is due to the capsid’s preference towards different sugars and receptors (Table 1). Internalization occurs by endocytosis, the CLIC/GEEC (clathrin-independent carriers/GPI enriched endocytic compartment) pathway or micropinocytosis, a process that is rapid (for AAV2 for example, it can be only 30 minutes). This is followed by retrograde transport along the endosomal pathway, where the capsid is modified by the low pH and enzymes, exposing VP1 and VP2 N-termini. The viral particle escapes the endosomal compartments and undergoes nuclear import through the nuclear pore complex (NPC), being directed by the basic residue clusters of VP1 and VP2. In the last steps of the process, the capsid is uncoated and using the host’s DNA polymerase double-stranded DNA is formed (Figure 1) (2). Our understanding of AAV transduction and efficiency has advanced considerably, but these viruses still maintain some of their mystery.
Table 1: AAV serotype receptors and tropism (based on reference 2).
| AAV Serotype | Glycan Receptors | Protein Receptors | Tropism |
|---|---|---|---|
| 1 | N-linked Sialic Acid | - | SM CNS Retina Pancreas |
| 2 | Heparan Sulfate Proteoglycan (HSPG) | FGFR1 HGFR LamR CD9 Tetraspanin |
Vascular Smooth Muscle Cells SM CNS Liver Kidney |
| 3 | HSPG | FGFR1 HGFR LamR |
SM |
| 4 | O-linked Sialic Acid | - | CNS Retina |
| 5 | N-linked Sialic Acid | PDGFR | SM CNS Retina Lung |
| 6 | N-linked Sialic Acid HSPG |
EGFR | SM Heart Lung |
| 7 | - | - | SM Retina CNS |
| 8 | - | LamR | Liver SM CNS Retinal Pancreas Heart |
| 9 | N-linked Galactose | LamR | Liver Heart Brain SM Lung Pancreas Kidney |
Abbreviations: Skelestal Muscle (SM), Central Nervous System (CNS), Fibroblast growth factor receptor 1 (FGFR1), HGFR (hepatocyte growth factor receptor), laminin receptor (LamR), Platelet-derived growth factor receptor (PDGFR), Epidermal growth factor receptor (EGFR)
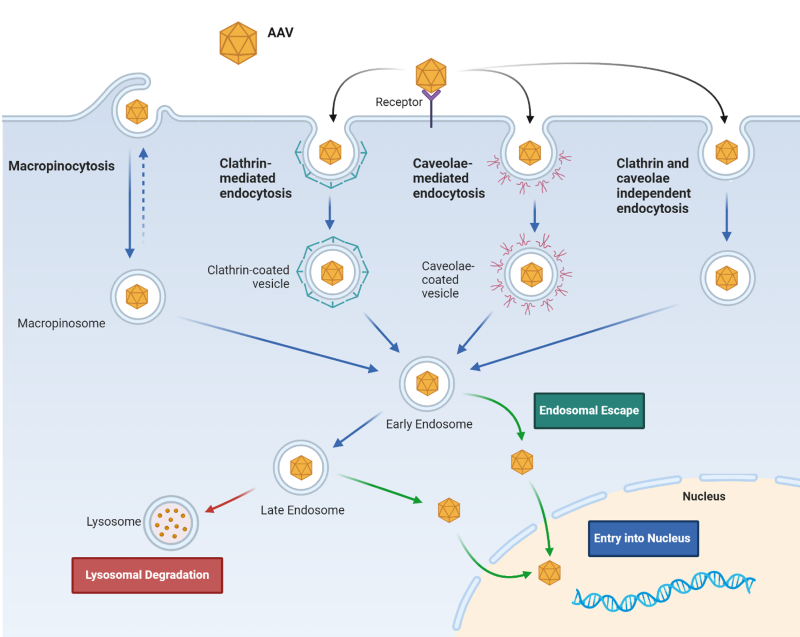
Figure 1: AAV cellular internalization.
Alteration of the tropism has been attempted via insertion of peptide ligands for cellular receptors that are non-native, insertion of random peptide sequences into specific regions, and by taking advantage of phenotypic variation by selecting variants created by random gene shuffling or homologous recombination. For example, AAV-DJ was selected from a library of AAV hybrids for its ability to enter a non-permissive human hepatoma cell line and for its resistance to pooled human serum (IVIG), which has a high neutralizing capacity against prevalent AAV serotypes. AAV-DJ is an AAV2/8/9 chimera, combining the broad in vitro tropism and specific liver in vivo tropism gained from AAV2, with the AAV8/9 ability to evade neutralization (3).
AAV engineering to create a genetic modification tool
Our current ability to perform genetic studies and link specific genes to a disease has allowed the gene therapy field to progress rapidly. Gene therapy refers to the administration of a normal gene in the case of recessive mutations or when the gene is absent, or the ability to normalize levels and activity at the molecular level, for instance by silencing a gene, and relies on the ability to safely deliver DNA to cells. Replacement of the AAV viral genome for ITR-flanked transgenes creates a recombinant AAV (rAAV), which has become the delivery method of choice in gene therapy.
rAAVs exist in the nucleus as episomes, and contrary to lentiviruses they are unable to integrate into the cellular genome. While this brings safety benefits, as there is no risk of random integration and activation of oncogenes, the cell division rate dictates the timeframe for transgene loss. Their application is thus particularly useful for the delivery of transgenes smaller than 5 kb to post-mitotic tissues like the brain, retinal, liver, skeletal muscle, and the heart (2). Attempts to fit larger transgenes into rAAV have resulted in interesting results, as the transgene seems to somehow get truncated and recombine, and in some cases create a functional protein (5). In the case of the protein dystrophin, a protein encoded by a 11.5 kb gene that when mutated results in Duchenne Muscular Dystrophy, attempts have been made to shrink the DNA to the minimal regions needed and create a mini dystrophin (6). The use of dual particles containing overlapping transgenes is another way to bypass the transgene size limitation when the exact domains necessary for a protein to be functional in the context of the disease are unknown.
The recombinant viral genome of modern vectors often includes the AAV2 ITRs, a mammalian promoter, the transgene of interest, and terminator. The most used promoters include CMV (cytomegalovirus), EF1a (elongation factor 1a), SV40 (simian virus 40), chicken β-actin and CAG (CMV, chicken β-actin, rabbit β-globin) (1). However, the use of tissue-specific promoters, such as the enolase promoter for neuron-specific expression, or DNA regulatory elements, avoids undesired transgene expression even if performing local AAV delivery. The choice of terminator/polyadenylation signals and the addition of mRNA stability elements or post-translational regulator elements can also have an impact on protein expression. The mutation of residues can increase transduction efficiencies by reducing the sensitivity to neutralizing antibodies or reducing proteolytic degradation once inside the cell. For instance, mutation of surface exposed tyrosine residues to phenylalanine prevents phosphorylation, ubiquitination, and degradation (1), and has been shown to increase transduction efficiency by 10- to 100-fold and transduction of nonpermissive cells (2). Additionally, the use of compounds such as doxorubicin and hydroxyurea, can enhance transduction by acting at different steps of the transduction pathway (2).
Nature has thus created a tool that seems almost perfectly designed for delivery of transgenes to post-mitotic cells. However, as discussed next, there are still challenges that need to be surpassed for their widespread application in gene therapy.
AAV Manufacturing Challenges
One of the biggest challenges companies and patients face when considering AAV-based therapies is the high manufacturing cost. New approaches are thus needed that will allow scientists to convert these tools from niche into widely applicable therapies.
The production of rAAV follows a workflow similar to lentiviruses, and some of the same handicaps. It typically involves the design and preparation of three plasmids, one encoding the gene of interest, one with the rep/cap genes, and a third one with the helper genes. It also requires a producer cell line, such as HEK293 cells. Introducing three plasmids into a cell and being able to scale it up are a challenge, resulting in a large fraction of viral particles not containing the transgene, which in turn requires appropriate quality control methods to distinguish empty capsids from the complete particles.
The use of adherent cells as producer cells is hard to scale up, and handling multiple vessels increases the risk of contamination and inconsistencies. One the other hand, the transfection of HEK293 cells in suspension is not as efficient and can result in the production of a larger fraction of empty capsids. Finally, the generation of stable cell lines has been limited in success. The introduction of helper genes is only possible for adenoviral E1a and E1b, but in HEK293 these lead to expression of rep, which is toxic to the cells. One alternative to HEK293 cells is the use of insect cells (Sf9 cells). These can be infected with 2-3 baculoviruses or integrate stably the rep/cap gene under the control of a promoter that is activated by infection with the baculovirus with the ITR-flanked gene of interest. Insect cells can reach very high cell densities in serum-free media, resulting up to 1015 GC (genome copies)/L, and baculoviruses are unable to transduce mammalian cells. Small scale production can be performed in shaker flasks, or Hyperflasks, whereas scaling makes use of WAVE or perfusion systems (1, 8). A few days after transfection, AAVs can be collected after cell lysis, but care is needed not to induce AAV aggregation or use compounds that can lead to toxicity in patients. Triton X-100 for instance has been classified as "of high concern" by the European Chemicals Agency. Purification and polishing are the next steps in the process and aim to remove cell debris and empty capsids present. The use of density ultracentrifugation with CsCl or iodixanol is usually sufficient for small scale but is not scalable and raises concerns about their impact on the properties of the particles. One alternative is the use of affinity chromatography with an AAV-specific binding protein (1). Finally, appropriate methods are needed to ensure the purity and quality of the viral particles and can include both biochemical and cellular based assays (7).
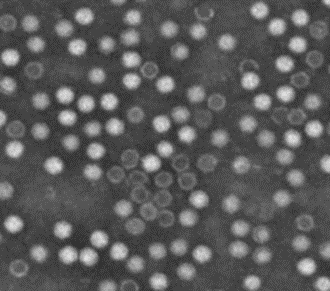
Figure 2: Electron microscopy characterization of AAV purity, showing the typical icosahedral particles. The lack of penetration of the dye indicates intact particles (from reference 7).
AAV in the clinic
There are more than 7,000 known rare diseases, each individually affecting a small portion of the population but that collectively account for 30 million Americans, two thirds being children (8). In 2012 FDA approved Glybera, an AAV-mediated gene therapy for LPLD (lipoprotein lipidase deficiency). As of 2023 there are 3 FDA-approved therapies, Luxturna for LCA (Leber’s congenital amaurosis), Zolgensma for SMA (spinal muscular dystrophy) and Hemgenix for Hemophilia B, with a price ranging from 850 thousand dollars to 3.5 million dollars (9). This high price tag and the low number of patients sadly resulted in the removal of Glybera from the market in 2018 (8), exemplifying the real need to overcome manufacturing and development costs.
In addition to their biology, the appeal to use rAAV in GT arises from their lower immunogenicity compared to other viruses. However, they are not fully non-immunogenic, and several aspects must be considered when designing a clinical study. Capsid proteins can trigger immune responses when delivered at high doses, with a response being generated until the proteins are degraded. The transgene itself can also cause immune responses, as in the case where the mutation resulted in no protein being expressed, and thus the immune system has never been exposed and adapted to that protein, or when an engineered sequence is recognized as foreign. Cells can also generate stress responses to an overexpressed protein and promote immune reactions. Most of us have at some point been exposed to AAV and have an adaptive response in place. In fact, this has been seen as one of main factors behind early clinical failures. Patients thus should be prescreened for NAbs (Neutralizing antibodies) against specific AAV variants, and alternatives can include the use of variants resistant to NAbs and the choice of a route of administration that bypasses the circulating immune responses (1, 10). AAVs are resistant to a broad range of temperature and pH, thus allowing several delivery routes to be used (1). The development of an immune response correlates with the site of injection and dosage. Even in immune-privileged environments, the delivery of very high doses of AAV can lead to inflammation, particularly if the environment is prone to inflammatory responses, and hepatotoxicity has been observed in 1/3 of the patients treated with Onasemnogene aberparvovec (10). The use of immunosuppressive drugs seems a viable option but requires an understanding of the exact trigger of the response for an appropriate choice of drug. A better understanding of the pathways activated in response to AAV treatment, and what triggers them, will be crucial for better treatments. Until then it may be wise to shift from delivering high dosages to the use of more efficient AAV in terms of transduction efficiency, transgene activity and expression, with new strategies including “coupled immunomodulation” where short TLR9 (toll-like receptor 9)- inhibitory sequences are included in the AAV genome (11). While these aspects should not be neglected, one should keep in perspective the benefit that can be gained from using AAV for GT.
One field that has greatly benefited from the use of AAV to cure genetic disorders is ophthalmology. The retina is an immune-privileged, self-contained, external organ, composed of terminally differentiated cells, characteristics that made it attractive for AAV GT. There are at least three clinical trials involving delivery of RPE65 to patients with LCA, one delivering REP1 (Rab escort protein 1) to choroideremia patients, two delivering genes in ACHM (achromatopsia), one for LHON (Leber’s hereditary optic neuropathy) and one for XLRS (X-linked retinoschisis syndrome) patients.
The next set of clinical trials targets hemophilia A and B, with systemic delivery of FVIII (factor VIII) and FIX (Factor IX) being attempted by multiple companies. Neurological disorders, where a direct administration into the CNS can be bypassed using AAV serotypes able to cross the BBB (blood-brain barrier) as in the case of plaque-specific antibodies delivery to Alzheimer disease (AD) patients, cardiac and lung diseases are also being studied and interestingly, aerosolized AAV can result in delivery of AAV to the lungs (see reference 1 for details on ongoing clinical trials).
BPS Bioscience AAV Research Solutions

Figure 3: mCherry expression and luciferase activity in HEK293 cells transduced with AAV2 Luciferase-mCherry (#78471).
HEK293 cells were transduced with AAV2 Luciferase mCherry particles and mCherry expression was assessed by fluorescent microscopy (left panel). Luciferase expression was determined using One-STEP™ Luciferase Assay System (#60690), using non-transduced HEK293 cells as control (right panel).
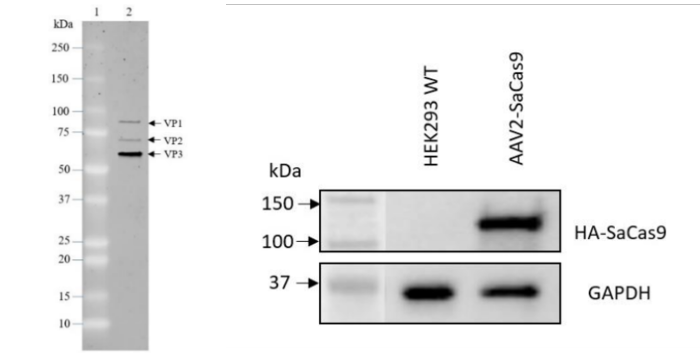
Figure 4. Quality control results for AAV2 SaCas9 (#78480).
AAV2 SaCas9 particles were analyzed by SDS-PAGE and confirmed to be 90% pure of contaminants (left panel). Expression of SaCas9 in HEK293 cells transduced with AAV2 SaCas9 was verified by Western Blot. GAPDH was used as loading control (right panel).
Our off-the-shelf products exemplify BPS Bioscience capability and know-how and can easily be applied to the development of custom AAV particles, expressing any protein of interest. The challenges associated with AAV manufacturing and time-consuming processes involved can be left in our hands, saving researchers time and costs.
Conclusions
rAAV has been shown to be safe in a broad number of clinical settings and diseases, making it a major tool in gene therapy. Studies into the molecular mechanisms involved in AAV transduction are still needed, and any new discovery can have a great impact on their clinical success. Strategies to enhance their efficiency, minimize immune responses, control transgene expression levels, make them applicable to larger genes and bypass the need to use helper virus during manufacture are still active fields of research. BPS Bioscience provides researchers with a set of tools, covering the main serotypes and typically used fluorescent proteins, plus luciferase and even Cas9, that can support new discoveries.
References
1. Naso M. et al., 2017 BioDrugs 31(4): 317-334.
2. Nonenmacher M. and Weber T., 2012 Gene Ther. 19(6): 649-658.
3. Lerch M., et al., 2012 Structure 20(8):1310-1320.
4. Liu Y., et al., 2015 Current Pharmaceutical Design 21(23):1-9.
5. Lopes VS., et al., 2013, Gene Ther 20: 824-833.
6. Elangkovan N. and Dickson G., 2021 J Neuromuscul Dis 8(Suppl2): S303-S316.
7. Allay J., et al., 2011 Human Gene Therapy 22:595-604.
8. Mendell J., et al., 2021 Mol Ther 29(2): 464-488.
9. Collins L., et al., 2023 ACS Synth Biol 12(1):17-26.