Emerging Therapeutic Approaches Against RAS Cancer Mutations
Introduction
RAS (rat sarcoma virus) proteins are small GTPases involved in signal transduction as part of the RAS/MAPK (mitogen-activated protein kinase) pathway and are involved in cell proliferation and differentiation. There are three human RAS genes: KRAS (Kristen rat sarcoma virus), HRAS (Harvey rat sarcoma virus) and NRAS (neuroblastoma rat sarcoma virus). The KRAS gene encodes two isoforms, KRAS-4B and KRAS-4A, but due to the high levels of KRAS-4B mRNA found in cells the term KRAS tends to generally refer to this isoform. Excessive activation of any of the RAS proteins is responsible for 20-25% of all cancers, with KRAS accounting for about 85% of all RAS mutations (1, 2). Mutations in RAS proteins also increase the chance of resistance to treatment. The ability to identify cancer-driving genes, such as RAS, and target them specifically has revolutionized the cancer therapy field. The high incidence and poor prognosis of KRAS mutations made them a prime target in drug development. In this technical note we will introduce RAS proteins, their role in disease and potential therapeutical avenues being pursued to target KRAS-driven cancer.
RAS proteins
RAS proteins belong to the GTPase family and are comprised of a catalytic domain responsible for guanine nucleotide binding, and a hypervariable region containing a CAAX motif that is farnesylated or prenylated, allowing specific membrane insertion and dictating the protein’s function. RAS proteins are involved in cell proliferation, differentiation, migration, apoptosis and metabolism. KRAS also participates in regulating viral immunity, endocytosis, membrane trafficking, adhesion, amongst others, through the variety of partners it interacts with and activates (1).
GTPases cycle between a GDP-bound (inactive) and a GTP-bound (active) state, in a process that involves two types of regulatory proteins: GEFs (guanine exchange factors) and GAPs (GTPase activating proteins). While at rest, RAS binds GDP and is inactive. KRAS can be activated indirectly by growth factors, chemokines, Ca2+ or tyrosine kinases). The binding of a growth factor to its receptor, such as EGF (epidermal growth factor) to EGFR (epidermal growth factor receptor), triggers a signaling cascade that results in activation of RAS by its respective GEF. There are three main Ras-GEF families, each one with specific roles. SOS (son of sevenless) tends to be involved in signaling mediated by RTKs, RAS-GRF regulates KRAS in a Ca2+ influx-calmodulin dependent mode (mainly in the brain), and RAS-GRP works via non-RTKs. The exchange of GDP for GTP triggers a conformational change that allows KRAS to bind its effectors and regulate signaling pathways, such as the RAF (rapidly accelerated fibrosarcoma) – MEK (mitogen activated protein kinase) – ERK (extracellular regulated kinase) or the PI3K (phosphoinositide 3- kinase)- AKT (protein kinase B) -mTOR (mammalian target of rapamycin) signaling pathways. In contrast, GAP such as neurofibromin 1 (NF1) promote the hydrolysis of GTP by RAS, completing the cycle (1, 3, 4) (Figure 1).
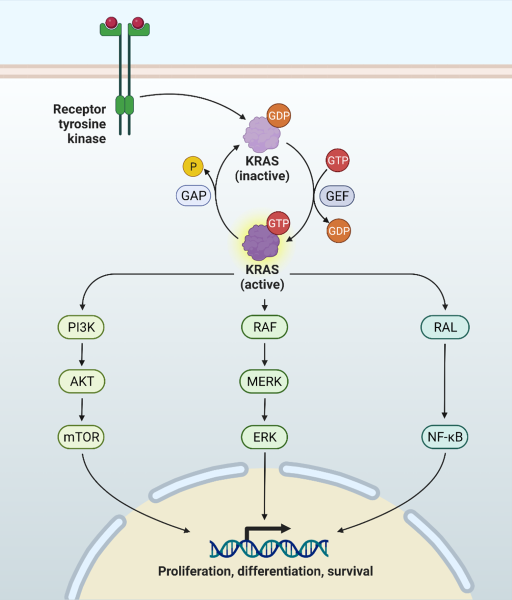
Figure 1: RAS cycle and RAS-regulated signaling pathways. RAS proteins cycle between an active GTP-bound state and an inactive GDP-bound state. GEF proteins replace GDP for GTP, while GAP proteins enhance the GTPase activity of RAS. In the active state RAS regulates several pathways, involved in multiple critical cellular functions.
KRAS mutations and cancer
As mentioned, KRAS accounts for 85% of all RAS family mutations, and it is considered the most common driver in human cancer. Examples include PDAC (pancreatic ductal adenocarcinoma), CRC (colorectal carcinoma) and NSCLC (non-small cell lung cancer). Single-base missense mutations are the most predominant abnormality found, and 98% of these can be found in 3 specific codons: G12, G13 and Q61. The codon and frequency of the mutation relate to tissue type, with 67.6% of PDAC being the result of KRAS mutations, and the G12D mutation being the main responsible. G12C is found frequently in NSCLC, where KRAs mutations account for 20.4% of all patients (1, 4). The incidence tends to be higher in smokers, thus smoking has been considered an important factor. The numbers are quite striking and provide clear evidence on how one single protein, undergoing a few specific mutations, can be responsible for such catastrophic outcomes (Table 1).
Table 1: Relation between RAS mutations and cancer type (2).
| Cancer Type | % due to RAS Mutations |
|---|---|
| AML (Acute Myeloid Leukemia) | 15% (5% due to KRAS mutations) |
| ALL (Acute Lymphoblastic Leukemia) | 11% |
| MM (Multiple Myeloma) | 30-40% (23% due to KRAS mutations) |
| BCP-ALL (B-Cell Precursor ALL) | 44% |
| CRC | 45% |
| Ovarian Cancer | 16-44% (all due to KRAS mutations) |
| Endometrial Cancer | 10-30% (all due to KRAS mutations) |
| Lung Adenocarcinoma | 40% (all due to KRAS mutations) |
| PDAC | 90% (all due to KRAS mutations) |
| Prostate Cancer | 7% (all due to KRAS mutations) |
| Melanoma | 1.7% (all due to KRAS mutations) |
An understanding of KRAS functions and how those functions are impacted by the predominantly encountered mutations is crucial for the development of appropriate therapeutic approaches. Mutations can impact GTPase activity, binding to effectors and location of the protein. The KRAS G12 mutations seem to make the protein insensitive to neurofibromin 1 GAP activity, with G12C and G12V-mutated cells having lower levels of phosphorylated AKT, but G12D mutation resulting in higher levels. G12R, G12D and G12V mutations lead to low affinity towards RAF, while G12A, Q61H and Q61L increase that affinity. A single mutation can thus result in multiple complex scenarios which determine the sensitivity of the mutation to targeted therapy and the clinical outcome. For example, patients with G12C and G12D mutations are prone to bone metastases, while patients with G12V often have pleuropericardial metastases (1).
Mutant KRAS tend to result in metabolic changes in tumor cells that favor cancer progression, such as upregulation of GLUT1 (glucose transporter 1) and hexokinase 1 and 2, increase in glucose uptake and glycolytic activity, formation of precursors of lipids, proteins and DNA, promotion of alternative glutamine metabolism leading to increased levels of NADPH, and autophagy (1, 3). In addition, KRAS mutations can create an inflammatory TME (tumor microenvironment) by mediating several pathways that result in high levels of inflammatory cytokine or chemokines. They can also mediate tumor immune escape by increasing PD-L1 (programmed death-ligand 1) mRNA stability, upregulating PD-L1 levels, downregulating MHC class I and pushing immune cells to become inhibitory (Figure 2). For example, CD4+ cells in the TME can differentiate into immunosuppressive Tregs in response to cytokine secretion driven by KRAS linked signaling pathways (1). The high metabolism exhibited by KRAS-driven cancer is also its Achille’s heel. Inhibition of glutamine degradation and autophagy, which work to feed cancer cells, has shown promising results in pre-clinical studies. Although more studies are needed, it is a path worth exploring.
It was found that in a population of 1078 NSCLC patients, 53% of the patients had co-mutations in other proteins, with 39.4% of those being on p53. A comparison of the TME in patients with KRAS/STK11 (serine/threonine kinase 11) and KRAS/p53 co-mutations has shown different TIL (tumor infiltrating lymphocytes) and Treg populations and different responses to PD-1 inhibitors. It is thus critical to have a deeper understanding of the exact KRAS mutation, cellular context and also potentially relevant co-mutations so the choice of treatment option can lead to a successful outcome.
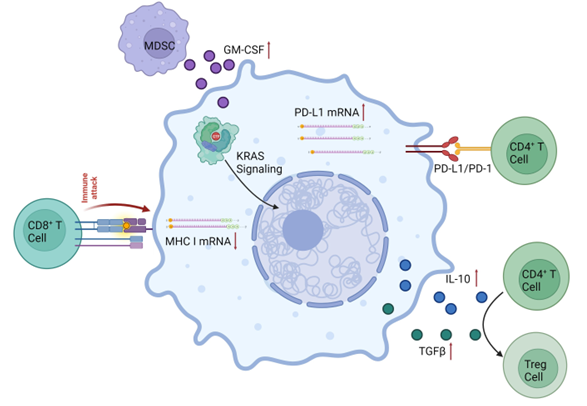
Figure 2: KRAS mutant contributions to the development of an immunosuppressive TME in KRAS-driven cancer. KRAS-mediated signaling leads to an increase in PD-L1 mRNA and protein levels. There is a decrease in MHC class I mRNA levels, creating a decrease in the immune response from CD8+ cytotoxic T cells. An increase in secretion of IL-10 and TGFβ leads to the differentiation of naïve CD4+ cells to Tregs, while an increase in levels of GM-CSF increases the presence of MDSCs (myeloid-derived suppressor cells). Adapted from (1).
Therapeutic approaches for KRAS-linked cancer
After 40 years of investigation, the puzzle of how to target KRAS mutants is still far from being complete. KRAS are small proteins, with a smooth almost spherical surface, and the only available protein pocket is the high affinity GTP binding pocket (GTP affinity at pM level) (3), and these characteristics have made it undruggable for many years. In recent years new therapeutical approaches have been developed and have resulted in promising results.
Some of the approaches to target mutant KRAS have been indirect strategies, aiming at reducing its levels, its localization, its interaction with effectors or synthetic lethality approaches with CDK (cyclin-dependent-kinase) inhibitors. These have resulted in modest or no benefit. For instance, the use of exosomes to deliver siRNA or antisense oligonucleotides in lung cancer or targeting farnesyltransferases and geranylgeranyl transferase type 1, and thus attempt to block delivery of KRAS to the cell membrane, has proved inefficient. Blocking the interaction between KRAS and its upstream and downstream effectors has been the focus of many studies, but it was found that inhibition of one pathway can result in stimulation of compensatory mechanisms making it necessary to use combinatory therapy of multiple inhibitors. In AML, inhibition of SOS1 with compounds such as BI-3406 seems to increase the sensitivity of KRAS-mutant cells to MEK inhibition, offering an example of combinatory therapy (2).
KRAS G12C-driven cancer is so far the most treatable KRAS mutant. The G12C mutant has a normal GTPase cycle and still allows interaction with effector proteins. Modifications of GDP, such as in the compound SML-8-73-1, that allow the GDP to bind covalently to Cys12 are one option to lock the protein in an inactive state. AMG510 (Sotorasib) was the first small molecule inhibitor to go into clinical trials and it has been approved in 2021 for the treatment of NSCLC with G12C mutations. Interestingly it seems that the main mechanism behind the potency of G12C inhibitors links not to their high affinity to the mutant protein but to the speed with which this mutant hydrolyzes GTP. Several clinical trials are ongoing using either specific KRAS G12C mutant inhibitors alone or in combination with other drugs. The use of targeted degradation by proteolysis-targeting chimaeras (PROTACs) and cancer vaccines have also been explored. The PROTAC LC-2, which uses VHL as E3 ligase has resulted in promising results, but further refinement is necessary so the PROTAC can undergo several cycles of KRAS degradation.
The development of therapies that target KRAS G12C mutant protein has been without a doubt a major scientific breakthrough that benefits patients daily. Unfortunately, newly synthesized mutant KRAS can still get activated via compensatory mechanisms and drug resistance can quickly occur, with treatments no longer being beneficial. The path forward seems to be the use of combination therapy. Drug resistance seems to be due in part to RTKs, opening the door to the use of inhibitors towards RTK and G12C, but it is of note that a synergistic effect may not be observed across different cancers, due to the heterogeneity of RTKs. Other approaches include combining inhibition of KRAS with points of pathway convergence, such as SHP2 (scr homology region 2 domain-containing phosphatase 2), targeting cell cycle checkpoints by inhibition of CDK4/6, or targeting immune checkpoints as with inhibitors of PD-1/PD-L1 (1, 4).
The development of approaches targeting other KRAS mutations, which do not have the ability to form a covalent binding to cysteines or that do not have high GTPase activity has not been highly successful, but progress has been made. MRTX1133 has been identified as a non-covalent inhibitor of KRAS G12D, by inhibiting the interaction of KRAS with effector proteins (4), but further studies are required.
Wild type KRAS can still contribute to oncogenesis, as in the case of breast cancer, where HER2 and EGFR are typically overexpressed, and result in RAS signal amplification. Melanoma is a cancer type where KRAS mutations have low incidence, and BRAF (v-Raf murine sarcoma viral oncogene homolog B) mutations are the main culprit. However, it was observed that inhibition of BRAF alone is only effective in a fraction of the patients, with KRAS inhibition acting synergistically in those cases.
The use of personalized medicine opens a new future in oncology, where the development of new potent specific KRAS inhibitors is crucial for combinatory therapy of cancers, KRAS-mutant or not.
Conclusions
KRAS is the major player in the most common cancer types and contributes to oncogenesis in multiple ways. Even when not directly involved or mutated, it can also contribute extensively to the development of disease. Despite several attempts and approaches tested, and many years of research, KRAS mutants remain an unsolved challenge in cancer therapy. The staggering number of cancers and number of patients that present KRAS mutations gives researchers no other option than to persevere in advancing the field and finding new and effective ways to target KRAS mutants, either directly or indirectly. BPS Bioscience can assist researchers in this challenging task with its high purity recombinant proteins, including GDP and GppNHp-loaded KRAS mutants and effectors, and assay kits specifically designed to assess KRAS nucleotide exchange activity. Our large range of products, including model cell lines, allows the study of the complex network of pathways linked to KRAS and makes BPS Bioscience an ideal partner in the field.
References
(1) Huang L., et al., 2021 Signal Transduction and Targeted Therapy 6: 386.
(2) Mustachio L., et al., 2021 Cancers (Basel) 13 (6): 1204.
(3) Beganoyic S., 2009 Bosn J Basic Med Sci 9(1): S17-S20.